Isovaleric acidemia
| Isovaleric acidemia | |
|---|---|
 | |
| Isovaleric acid | |
| Classification and external resources | |
| Specialty | endocrinology |
| ICD-10 | E71.1 |
| ICD-9-CM | 270.3 |
| OMIM | 243500 |
| DiseasesDB | 29840 |
Isovaleric acidemia, also called isovaleric aciduria or isovaleric acid CoA dehydrogenase deficiency,[1] is a rare autosomal recessive[2] metabolic disorder which disrupts or prevents normal metabolism of the branched-chain amino acid leucine. It is a classical type of organic acidemia.[3]
Symptoms
A characteristic feature of isovaleric acidemia is a distinctive odor of sweaty feet.[4] This odor is caused by the buildup of a compound called isovaleric acid in affected individuals.
In about half of cases, the signs and symptoms of this disorder become apparent within a few days after birth and include poor feeding, vomiting, seizures, and lack of energy that can progress to coma. These medical problems are typically severe and can be life-threatening. In the other half of cases, the signs and symptoms of the disorder appear during childhood and may come and go over time. They are often triggered by an infection or by eating an increased amount of protein-rich foods.
Diagnosis
The urine of newborns can be screened for isovaleric acidemia using mass spectrometry,[3] allowing for early diagnosis. Elevations of isovalerylglycine in urine and of isovalerylcarnitine in plasma are found.
Genetics
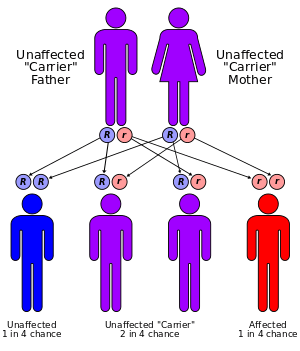
The disorder has an autosomal recessive inheritance pattern, which means the defective gene is located on an autosome, and two copies of the gene - one from each parent - must be inherited to be affected by the disorder. The parents of a child with an autosomal recessive disorder are carriers of one copy of the defective gene, but are usually not affected by the disorder.
Mutations in both copies of the IVD gene result in isovaleric acidemia.
Pathophysiology
The enzyme encoded by IVD, isovaleric acid-CoA dehydrogenase (EC 1.3.99.10), plays an essential role in breaking down proteins from the diet. Specifically, the enzyme is responsible for the third step in processing leucine, an essential amino acid. If a mutation in the IVD gene reduces or eliminates the activity of this enzyme, the body is unable to break down leucine properly. As a result, isovaleric acid and related compounds build up to toxic levels, damaging the brain and nervous system.
Treatment
Treatment consists of dietary protein restriction, particularly leucine. During acute episodes, glycine is sometimes given, which conjugates with isovalerate forming isovalerylglycine, or carnitine which has a similar effect.
Elevated 3-hydroxyisovaleric acid is a clinical biomarker of biotin deficiency. Without biotin, leucine and isoleucine cannot be metabolized normally and results in elevated synthesis of isovaleric acid and consequently 3-hydroxyisovaleric acid, isovalerylglycine, and other isovaleric acid metabolites as well. Elevated serum 3-hydroxyisovaleric acid concentrations can be caused by supplementation with 3-hydroxyisovaleric acid, genetic conditions, or dietary deficiency of biotin. Some patients with isovaleric acidemia may benefit from supplemental biotin.[5] Biotin deficiency on its own can have severe physiological and cognitive consequences[6] that closely resemble symptoms of organic acidemias.
Prognosis
A 2011 review of 176 cases found that early diagnosis (within a few days of birth) was associated with a mortality of 33%. For the late diagnosis group, mortality was only 3%.[7]
Epidemiology
Isovaleric acidemia is estimated to affect at least 1 in 250,000 births in the United States.[8]
Screening
On 9 May 2014, the UK National Screening Committee (UK NSC) announced its recommendation to screen every newborn baby in the UK for four further genetic disorders as part of its NHS Newborn Blood Spot Screening programme, including isovaleric acidemia.[9]
See also
References
- ↑ Online Mendelian Inheritance in Man (OMIM) 243500
- ↑ Lee, Yw; Lee, Dh; Vockley, J; Kim, Nd; Lee, Yk; Ki, Cs (September 2007). "Different spectrum of mutations of isovaleryl-CoA dehydrogenase (IVD) gene in Korean patients with isovaleric acidemia". Molecular genetics and metabolism. 92 (1–2): 71–7. doi:10.1016/j.ymgme.2007.05.003. PMC 4136440
 . PMID 17576084.
. PMID 17576084. - 1 2 Dionisi-Vici, C; Deodato, F; Röschinger, W; Rhead, W; et al. (2006). "Classical organic acidurias, propionic aciduria, methylmalonic aciduria, and isovaleric aciduria: long-term outcome and effects of expanded newborn screening using tandem mass spectrometry". J Inherit Metab Dis. 29 (2–3): 383–389. doi:10.1007/s10545-006-0278-z. PMID 16763906.
- ↑ Tokatli A, Oskun T, Ozalp I (1998). "Isovaleric acidemia. Clinical presentation of 6 cases". The Turkish Journal of Pediatrics. 40 (1): 111–119. PMID 9673537.
- ↑ http://www.ommbid.com/OMMBID/the_online_metabolic_and_molecular_bases_of_inherited_disease/b/abstract/part9/ch93
- ↑ "Genova Diagnostics (GDX) - Diagnostic Laboratory Testing for Wellness & Preventive Medicine". metametrix.com.
- ↑ Grünert, Sarah C.; Wendel, Udo; Lindner, Martin; Leichsenring, Michael; Schwab, K. Otfried; Vockley, Jerry; Lehnert, Willy; Ensenauer, Regina (2012-01-01). "Clinical and neurocognitive outcome in symptomatic isovaleric acidemia". Orphanet Journal of Rare Diseases. 7: 9. doi:10.1186/1750-1172-7-9. ISSN 1750-1172. PMC 3292949. PMID 22277694.
- ↑ "Isovaleric acidemia". Genetics Home Reference. 4 May 2015.
- ↑ "New screening will protect babies from death and disability". screening.nhs.uk.
External links
- Organic Acidemia Association
- Isovaleric acidemia at NLM Genetics Home Reference
- -187695062 at GPnotebook
- GeneReviews: The Organic Acidemias