Philadelphia chromosome
| Philadelphia chromosome | |
|---|---|
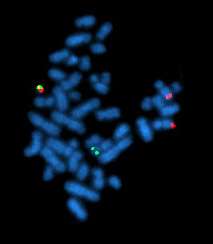 | |
| A metaphase cell positive for the bcr/abl rearrangement using FISH | |
| Classification and external resources | |
| Specialty | oncology |
| ICD-10 | C92.1 |
| ICD-9-CM | 205.1 |
| ICD-O | 9875/3 |
| MeSH | D010677 |
The Philadelphia chromosome or Philadelphia translocation is a specific genetic abnormality in chromosome 22 of leukemia cancer cells (particularly chronic myelogenous leukemia (CML) cells). This chromosome is defective and unusually short because of reciprocal translocation of genetic material between chromosome 9 and chromosome 22, and contains a fusion gene called BCR-ABL1. This gene is the ABL1 gene of chromosome 9 juxtaposed onto the BCR gene of chromosome 22, coding for a hybrid protein: a tyrosine kinase signalling protein that is "always on", causing the cell to divide uncontrollably.[1]
The presence of this translocation is a highly sensitive test for CML, since 95% of people with CML have this abnormality (the remainder have either a cryptic translocation that is invisible on G-banded chromosome preparations, or a variant translocation involving another chromosome or chromosomes as well as the long arm of chromosomes 9 and 22). However, the presence of the Philadelphia (Ph) chromosome is not sufficiently specific to diagnose CML, since it is also found in acute lymphoblastic leukemia[2] (ALL, 25–30% in adult and 2–10% in pediatric cases) and occasionally in acute myelogenous leukemia (AML).
Molecular biology
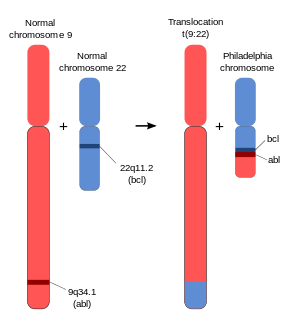
The chromosomal defect in the Philadelphia chromosome is a translocation, in which parts of two chromosomes, 9 and 22, swap places. The result is that a fusion gene is created by juxtapositioning the Abl1 gene on chromosome 9 (region q34) to a part of the BCR ("breakpoint cluster region") gene on chromosome 22 (region q11). This is a reciprocal translocation, creating an elongated chromosome 9 (termed a derivative chromosome, or der 9), and a truncated chromosome 22 (the Philadelphia chromosome).[3][4] In agreement with the International System for Human Cytogenetic Nomenclature (ISCN), this chromosomal translocation is designated as t(9;22)(q34;q11). Abl stands for "Abelson", the name of a leukemia virus which carries a similar protein.
Translocation results in an oncogenic BCR-ABL gene fusion that can be found on the shorter derivative 22 chromosome. This gene encodes for a Bcr-abl fusion protein. Depending on the precise location of fusion, the molecular weight of this protein can range from 185 to 210 kDa. Consequently, bcr-abl is referred to as p210 or p185.
Three clinically important variants are the p190, p210, and p230 isoforms.[5] p190 is generally associated with acute lymphoblastic leukemia (ALL), while p210 is generally associated with chronic myeloid leukemia but can also be associated with ALL.[6] p230 is usually associated with chronic myelogenous leukemia associated with neutrophilia and thrombocytosis.[6] Additionally, the p190 isoform can also be expressed as a splice variant of p210.[7]
The Abl gene expresses a membrane-associated protein, a tyrosine kinase, and the BCR-Abl transcript is also translated into a tyrosine kinase. The activity of tyrosine kinases is typically controlled by other molecules, but the mutant tyrosine kinase of the BCR-Abl transcript codes for a protein that is "always on" or continuously activated, which results in unregulated cell division (i.e. cancer). Although the BCR region also expresses serine/threonine kinases, the tyrosine kinase function is very relevant for drug therapy. Tyrosine kinase inhibitors (such as imatinib and sunitinib) are important drugs against a variety of cancers including CML, renal cell carcinoma (RCC) and gastrointestinal stromal tumors (GISTs).
The fused BCR-Abl protein interacts with the interleukin-3 receptor beta(c) subunit. The ABL tyrosine kinase activity of BCR-Abl is elevated relative to wild-type ABL.[8] Since ABL activates a number of cell cycle-controlling proteins and enzymes, the result of the BCR-Abl fusion is to speed up cell division. Moreover, it inhibits DNA repair, causing genomic instability and potentially causing the feared blast crisis in CML.
Nomenclature
The Philadelphia chromosome is designated Ph (or Ph') chromosome and designates the shortened chromosome 22. It arises from the translocation, which is termed t(9;22)(q34.1;q11.2). This means there is a translocation between chromosome 9 and chromosome 22, with breaks happening in region (3), band (4), sub-band (1) of the long arm (q) of chromosome 9 and region (1), band (1), sub-band (2) of the long arm (q) of chromosome 22. Hence the chromosome breakpoints are written as (9q34.1) and (22q11.2), respectively, using ISCN standards. Notation omitting the sub-bands is also commonly seen: t(9;22)(q34;q11).
Therapy
Tyrosine kinase inhibitors
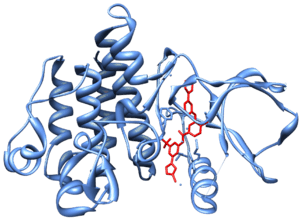
In the late 1990s, STI-571 (imatinib, Gleevec/Glivec) was identified by the pharmaceutical company Novartis (then known as Ciba Geigy) in high-throughput screens for tyrosine kinase inhibitors. Subsequent clinical trials led by Dr. Brian J. Druker at Oregon Health & Science University in collaboration with Dr. Charles Sawyers and Dr. Moshe Talpaz demonstrated that STI-571 inhibits proliferation of BCR-ABL-expressing hematopoietic cells. Although it did not eradicate CML cells, it did greatly limit the growth of the tumor clone and decreased the risk of the feared "blast crisis". In 2000 Dr. John Kuriyan determined the mechanism by which STI-571 inhibits the Abl kinase domain.[9] It was marketed in 2001 by Novartis as imatinib mesylate (Gleevec in the US, Glivec in Europe).
Other pharmacological inhibitors are being developed, which are more potent and/or are active against the emerging Gleevec/Glivec resistant BCR-abl clones in treated patients. The majority of these resistant clones are point-mutations in the kinase of BCR-abl. New inhibitors include dasatinib and nilotinib, which are significantly more potent than imatinib and may overcome resistance.
Axitinib, a drug used to treat renal cell carcinoma, has been shown to be effective at inhibiting the Abl kinase activity in patients with BCR-ABL1(T315I).[10] The T315I mutation in the fusion gene confers resistance to other tyrosine kinase inhibitors such as imatinib, however axitinib has been successfully been used to treat a patient with ALL carrying this mutation, as well as CML cells in culture.
Treatment of pediatric Ph+ ALL with a combination of standard chemotherapy and RTK inhibitors may result in remission, but the curative potential is unknown.
Blood or marrow transplants
A potentially curative, but risky, option for pediatric Ph+ ALL or Ph+ CML is bone marrow transplant or cord blood transplant, but chemotherapy is favored by some for achieving first remission (CR1). For some, bone marrow transplant from a matched sibling donor or a matched, unrelated donor may be favored when remission is obtained.
Cord blood transplant is favored by some when a 10/10 bone marrow match is not available, and cord blood transplant may have some advantages, including a reduced incidence of graft-vs-host disease (GVHD), which is a common and significant complication of transplant. However, transplant with cord blood sometimes requires longer periods of time for engraftment, which may increase the potential for complications due to infection. Regardless of the type of transplant, transplant-related mortality and relapse are possible, and the rates may change as treatment protocols improve. For second remission (CR2), if achieved, both chemotherapy and transplant options are possible, and many physicians prefer transplant.
History
The Philadelphia chromosome was first discovered and described in 1960 by Peter Nowell from the University of Pennsylvania School of Medicine and David Hungerford from the Institute for Cancer Research at the Lankenau Hospital Research Institute (now known as Lankenau Institute for Medical Research) and was therefore named after the city in which both facilities are located.[1][11][12]
In 1973, Janet D. Rowley at the University of Chicago identified the mechanism by which the Philadelphia chromosome arises as a translocation.[1][13][14]
See also
References
- 1 2 3 Wapner J.The Philadelphia Chromosome: A Genetic Mystery, a Lethal Cancer, and the Improbable Invention of a Lifesaving Treatment. ISBN 9781615191970
- ↑ Talpaz M, Shah NP, Kantarjian H, et al. (June 2006). "Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias". N. Engl. J. Med. 354 (24): 2531–41. doi:10.1056/NEJMoa055229. PMID 16775234.
- ↑ Kurzrock, R.; Kantarjian, H. M.; Druker, B. J.; Talpaz, M. (2003). "Philadelphia chromosome-positive leukemias: From basic mechanisms to molecular therapeutics". Annals of Internal Medicine. 138 (10): 819–830. doi:10.7326/0003-4819-138-10-200305200-00010. PMID 12755554.
- ↑ Melo, J. V. (1996). "The molecular biology of chronic myeloid leukaemia". Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, U.K. 10 (5): 751–756. PMID 8656667.
- ↑ Advani, A. S.; Pendergast, A. M. (2002). "Bcr-Abl variants: Biological and clinical aspects". Leukemia research. 26 (8): 713–720. doi:10.1016/s0145-2126(01)00197-7. PMID 12191565.
- 1 2 Pakakasama, S.; Kajanachumpol, S.; Kanjanapongkul, S.; Sirachainan, N.; Meekaewkunchorn, A.; Ningsanond, V.; Hongeng, S. (2008). "Simple multiplex RT-PCR for identifying common fusion transcripts in childhood acute leukemia". International Journal of Laboratory Hematology. 30 (4): 286–291. doi:10.1111/j.1751-553X.2007.00954.x. PMID 18665825.
- ↑ Lichty, B. D.; Keating, A.; Callum, J.; Yee, K.; Croxford, R.; Corpus, G.; Nwachukwu, B.; Kim, P.; Guo, J.; Kamel-Reid, S. (1998). "Expression of p210 and p190 BCR-ABL due to alternative splicing in chronic myelogenous leukaemia". British journal of haematology. 103 (3): 711–715. doi:10.1046/j.1365-2141.1998.01033.x. PMID 9858221.
- ↑ Sattler, Martin; James D. Griffin (April 2001). "Mechanisms of transformation by the BCR/ABL oncogene". International Journal of Hematology. 73 (3): 278–91. doi:10.1007/BF02981952. PMID 11345193.
- ↑ Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J (2000). "AStructural mechanism for STI-571 inhibition of abelson tyrosine kinase". Science. 289 (5486): 1857–9. doi:10.1126/science.289.5486.1938. PMID 10988075.
- ↑ Tea Pemovska; Eric Johnson; Mika Kontro; Gretchen A. Repasky; Jeffrey Chen; Peter Wells; Ciarán N. Cronin; Michele McTigue; Olli Kallioniemi; Kimmo Porkka; Brion W. Murray; Krister Wennerberg. "Axitinib effectively inhibits BCR-ABL1(T315I) with a distinct binding conformation". Nature. 519: 102–105. doi:10.1038/nature14119.
- ↑ Fox Chase Cancer Center. "50th Anniversary of the Discovery of the Philadelphia Chromosome".
- ↑ Nowell P, Hungerford D (November 1960). "A minute chromosome in chronic granulocytic leukemia" (PDF). Science. 132 (3438): 1497. doi:10.1126/science.132.3438.1488.
- ↑ Rowley JD (1973). "Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining". Nature. 243 (5405): 290–3. doi:10.1038/243290a0. PMID 4126434.
- ↑ Claudia Dreifus (2011-02-07). "The Matriarch of Modern Cancer Genetics". New York Times.
External links
- Philadelphia chromosome at the US National Library of Medicine Medical Subject Headings (MeSH)
- bcr-abl Fusion Proteins at the US National Library of Medicine Medical Subject Headings (MeSH)