Diffuse axonal injury
| Diffuse axonal injury | |
|---|---|
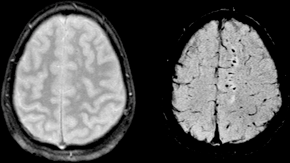 | |
| Susceptibility weighted image (SWI) of diffuse axonal injury in trauma at 1.5 teslas (right) | |
| Classification and external resources | |
| eMedicine | radio/216 |
| MeSH | D020833 |
Diffuse axonal injury (DAI) is a brain injury in which damage in the form of extensive lesions in white matter tracts occurs over a widespread area. DAI is one of the most common and devastating types of traumatic brain injury,[1] and is a major cause of unconsciousness and persistent vegetative state after severe head trauma.[2] It occurs in about half of all cases of severe head trauma and may be the primary damage that occurs in concussion. The outcome is frequently coma, with over 90% of patients with severe DAI never regaining consciousness.[2] Those who do wake up often remain significantly impaired.[3]
DAI can occur in every degree of severity from very mild or moderate to very severe.[4] Concussion may be a milder type of diffuse axonal injury.
Mechanism
Unlike brain trauma that occurs due to direct impact and deformation of the brain, DAI is the result of traumatic shearing forces that occur when the head is rapidly accelerated or decelerated, as may occur in car accidents, falls, and assaults.[5] It usually results from rotational forces or severe deceleration. Vehicle accidents are the most frequent cause of DAI; it can also occur as the result of child abuse[6] such as in shaken baby syndrome.[7]
The major cause of damage in DAI is the disruption of axons, the neural processes that allow one neuron to communicate with another. Tracts of axons, which appear white due to myelination, are referred to as white matter. Acceleration causes shearing injury: damage inflicted as tissue slides over other tissue. When the brain is accelerated, parts of differing densities and distances from the axis of rotation slide over each other, stretching axons that traverse junctions between areas of different density, especially at junctions between white and grey matter.[2] Two thirds of DAI lesions occur in areas where grey and white matter meet.[2]
Characteristics
Lesions typically exist in the white matter of brains injured by DAI; these lesions vary in size from about 1–15 mm and are distributed in a characteristic way.[2] DAI most commonly affects white matter in areas including the brain stem, the corpus callosum, and the cerebral hemispheres.
The lobes of the brain most likely to be injured are the frontal and temporal lobes.[8] Other common locations for DAI include the white matter in the cerebral cortex, the superior cerebral peduncles,[7] basal ganglia, thalamus, and deep hemispheric nuclei.[9] These areas may be more easily damaged because of the difference in density between them and the rest of the brain.[9]
Histological characteristics
DAI is characterized by axonal separation, in which the axon is torn at the site of stretch and the part distal to the tear degrades. While it was once thought that the main cause of axonal separation was tearing due to mechanical forces during the trauma, it is now understood that axons are not typically torn upon impact; rather, secondary biochemical cascades, which occur in response to the primary injury (which occurs as the result of mechanical forces at the moment of trauma) and take place hours to days after the initial injury, are largely responsible for the damage to axons.[5][10][11]
Though the processes involved in secondary brain injury are still poorly understood, it is now accepted that stretching of axons during injury causes physical disruption to and proteolytic degradation of the cytoskeleton.[1] It also opens sodium channels in the axolemma, which causes voltage-gated calcium channels to open and Ca2+ to flow into the cell.[1] The intracellular presence of Ca2+ unleashes several different pathways, including activating phospholipases and proteolytic enzymes, damaging mitochondria and the cytoskeleton, and activating secondary messengers, which can lead to separation of the axon and death of the cell.[5]
Cytoskeleton disruption

Axons are normally elastic, but when rapidly stretched they become brittle, and the axonal cytoskeleton can be broken. Misalignment of cytoskeletal elements after stretch injury can lead to tearing of the axon and death of the neuron. Axonal transport continues up to the point of the break in the cytoskeleton, but no further, leading to a buildup of transport products and local swelling at that point.[12] When it becomes large enough, swelling can tear the axon at the site of the break in the cytoskeleton, causing it to draw back toward the cell body and form a bulb.[4] This bulb is called a retraction ball, the hallmark of diffuse axonal injury.[2]
When the axon is transected, Wallerian degeneration, in which the part of the axon distal to the break degrades, takes place within one to two days after injury.[13] The axolemma disintegrates,[13] myelin breaks down and begins to detach from cells in an anterograde direction (from the body of the cell toward the end of the axon),[14] and nearby cells begin phagocytic activity, engulfing debris.[15]
Calcium influx
While sometimes only the cytoskeleton is disturbed, frequently disruption of the axolemma occurs as well, causing the influx of Ca2+ into the cell and unleashing a variety of degrading processes.[13][16] An increase in Ca2+ and Na+ levels and a drop in K+ levels is found within the axon directly after injury.[5][13] Possible routes of Ca2+ entry include sodium channels, pores torn in the membrane during stretch, and failure of ATP-dependent transporters due to mechanical blockage or lack of energy.[5] High levels of intracellular Ca2+, the major cause of post-injury cell damage,[17] destroy mitochondria,[4] and trigger phospholipases and proteolytic enzymes that damage Na+ channels and degrade or alter the cytoskeleton and the axoplasm.[18][13] Excess Ca2+ can also lead to damage to the blood brain barrier and swelling of the brain.[17]
One of the proteins activated by the presence of calcium in the cell is calpain, a Ca2+-dependent non-lysosomal protease.[18] About 15 minutes to half an hour after the onset of injury, a process called calpain-mediated spectrin proteolysis, or CMSP, begins to occur.[19] Calpain breaks down a molecule called spectrin, which holds the membrane onto the cytoskeleton, causing the formation of blebs and the breakdown of the cytoskeleton and the membrane, and ultimately the death of the cell.[18][19] Other molecules that can be degraded by calpains are microtubule subunits, microtubule-associated proteins, and neurofilaments.[18]
Generally occurring one to six hours into the process of post-stretch injury, the presence of calcium in the cell initiates the caspase cascade, a process in cell injury that usually leads to apoptosis, or "cell suicide".[19]
Mitochondria, dendrites, and parts of the cytoskeleton damaged in the injury have a limited ability to heal and regenerate, a process which occurs over 2 or more weeks.[20] After the injury, astrocytes can shrink, causing parts of the brain to atrophy.[2]
Diagnosis

DAI is difficult to detect since it does not show up well on CT scans or with other macroscopic imaging techniques, though it shows up microscopically.[2] However, there are characteristics typical of DAI that may or may not show up on a CT scan. Diffuse injury has more microscopic injury than macroscopic injury and is difficult to detect with CT and MRI, but its presence can be inferred when small bleeds are visible in the corpus callosum or the cerebral cortex.[21] MRI is more useful than CT for detecting characteristics of diffuse axonal injury in the subacute and chronic time frames.[22] Newer studies such as Diffusion Tensor Imaging are able to demonstrate the degree of white matter fiber tract injury even when the standard MRI is negative. Since axonal damage in DAI is largely a result of secondary biochemical cascades, it has a delayed onset, so a person with DAI who initially appears well may deteriorate later. Thus injury is frequently more severe than is realized, and medical professionals should suspect DAI in any patients whose CT scans appear normal but who have symptoms like unconsciousness.[2]
MRI is more sensitive than CT scans, but MRI may also miss DAI, because it identifies the injury using signs of edema, which may not be present.[20]
DAI is classified into grades based on severity of the injury. In Grade I, widespread axonal damage is present but no focal abnormalities are seen. In Grade II, damage found in Grade I is present in addition to focal abnormalities, especially in the corpus callosum. Grade III damage encompasses both Grades I and II plus rostral brain stem injury and often tears in the tissue.[23]
Treatment
DAI currently lacks a specific treatment beyond what is done for any type of head injury, including stabilizing the patient and trying to limit increases in intracranial pressure (ICP).
History
The idea of DAI first came about as a result of studies by Sabina Strich on lesions of the white matter of individuals who had suffered head trauma years before.[24] Strich first proposed the idea in 1956, calling it diffuse degeneration of white matter, however the more concise term "Diffuse Axonal Injury" was eventually preferred.[25] Strich was researching the relationship between dementia and head trauma[24] and asserted in 1956 that DAI played an integral role in the eventual development of dementia due to head trauma.[6] The term DAI was introduced in the early 1980s.[26]
Notable examples
- Top Gear presenter Richard Hammond suffered a DAI as a result of the Vampire Dragster Crash in 2006.
- Champ Car World Series driver Roberto Guerrero suffered a DAI as a result of a crash during testing at the Indianapolis Motor Speedway in 1987.[27]
- Formula 1 driver Jules Bianchi suffered a DAI as a result of an accident at the 2014 Japanese Grand Prix[28] and died, without regaining consciousness, 9 months later on 17 July 2015.[29]
See also
References
- 1 2 3 Iwata A., Stys P.K., Wolf J.A., Chen X.H., Taylor, A.G., Meaney D.F., and Smith D.H. (2004). Traumatic axonal injury induces proteolytic cleavage of the voltage-gated sodium channels modulated by tetrodotoxin and protease inhibitors. The Journal of Neuroscience. 24 (19): 4605—4613.
- 1 2 3 4 5 6 7 8 9 Wasserman J. and Koenigsberg R.A. (2007). Diffuse axonal injury. Emedicine.com. Retrieved on 2008-01-26.
- ↑ Vinas F.C. and Pilitsis J. (2006). Penetrating head trauma. Emedicine.com. Retrieved on 2008-01-14.
- 1 2 3 Smith, D.H. and Meaney D.F. (2000). Axonal damage in traumatic brain injury. The Neuroscientist. 6 (6): 483—495.
- 1 2 3 4 5 Wolf J.A., Stys P.K., Lusardi T., Meaney D., and Smith, D.H. (2001). Traumatic axonal injury induces calcium influx modulated by tetrodotoxin-sensitive sodium channels. Journal of Neuroscience. 21 (6): 1923–1930
- 1 2 Hardman JM, Manoukian A (2002). "Pathology of head trauma". Neuroimaging Clinics of North America. 12 (2): 175–187, vii. doi:10.1016/S1052-5149(02)00009-6. PMID 12391630.
- 1 2 Smith D. and Greenwald B. 2003.Management and staging of traumatic brain injury. Emedicine.com. Retrieved through web archive on 17 January 2008.
- ↑ Boon R. and de Montfor G.J. 2002. Brain injury. Learning Discoveries Psychological Services. Retrieved through web archive on 17 January 2008.
- 1 2 Singh J and Stock A (September 25, 2006). Head Trauma. Emedicine.com. Retrieved on 2008-01-17.
- ↑ Vulnerability of central neurons to secondary insults after in vitro mechanical stretch]. Journal of Neuroscience 24 (37): 8106—8123. PMID 15371512.
- 1 2 Mouzon, B; Chaytow, H (December 2012). "Repetitive Mild Traumatic Brain Injury in a Mouse Model Produces Learning and Memory Deficits Accompanied by Histological Changes.". J Neurotrauma. 29 (18): 2761–73. doi:10.1089/neu.2012.2498. PMID 22900595.
- ↑ Staal JA, Dickson TC, Chung RS, Vickers JC. (2007). Cyclosporin-A treatment attenuates delayed cytoskeletal alterations and secondary axotomy following mild axonal stretch injury. Dev Neurobiol. 67 (14): 1831-42. PMID 17702000.
- 1 2 3 4 5 LoPachin R.M. and Lehning E.J. (1997). Mechanism of calcium entry during axon injury and degeneration. Toxicology and Applied Pharmacology. 143 (2): 233—244. PMID 9144441.
- ↑ Cowie R.J. and Stanton G.B. (2005). Axoplasmic transport and neuronal responses to injury. Howard University College of Medicine. Retrieved on 2008-01-17.
- ↑ Hughes P.M., Wells G.M.A., Perry V.H., Brown M.C., and Miller K.M. (2002). Comparison of matrix metalloproteinase expression during wallerian degeneration in the central and peripheral nervous systems. Neuroscience. 113 (2): 273—287. PMID 12127085.
- ↑ Povlishock J.T. and Pettus E.H. (1996). Traumatically induced axonal damage: Evidence for enduring changes in axolemmal permeability with associated cytoskeletal change. Acta Neurochirurgica. 66: 81—86. PMID 8780803.
- 1 2 Zhou F., Xiang Z., Feng W.X., and Zhen L.X. (2001). Neuronal free Ca2+ and BBB permeability and ultrastructure in head injury with secondary insult. Journal of Clinical Neuroscience. 8 (6): 561—563. PMID 11683606.
- 1 2 3 4 Castillo M.R. and Babson J.R. (1998). Ca2+-dependent mechanisms of cell injury in cultured cortical neurons. Neuroscience. 86 (4): 1133—1144. PMID 9697120.
- 1 2 3 Büki A, Okonkwo D.O., Wang K.K.W., and Povlishock J.T. (2000). Cytochrome c release and caspase activation in traumatic axonal injury. Journal of Neuroscience. 20 (8): 2825—2834. PMID 10751434.
- 1 2 Corbo J. and Tripathi P. 2004. Delayed presentation of diffuse axonal injury: A case report. Trauma. 44 (1): 57–60. PMID 15226709.
- ↑ Crooks CY, Zumsteg JM, Bell KR (November 2007). "Traumatic brain injury: A review of practice management and recent advances". Phys Med Rehabil Clin N Am. 18 (4): 681–710. doi:10.1016/j.pmr.2007.06.005. PMID 17967360.
- ↑ Maas AI, Stocchetti N, Bullock R (August 2008). "Moderate and severe traumatic brain injury in adults". Lancet Neurology. 7 (8): 728–41. doi:10.1016/S1474-4422(08)70164-9. PMID 18635021.
- ↑ Bigler, E.D. 2000. The Lesion(s) in Traumatic brain injury: Implications for clinical neuropsychology. Retrieved through web archive on 17 January 2008.
- 1 2 Pearce JM (2007). "Observations on concussion. A review". European Neurology. 59 (3–4): 113–119. doi:10.1159/000111872. PMID 18057896.
- ↑ Gennarelli GA, Graham DI (2005). "Neuropathology". In Silver JM, McAllister TW, Yudofsky SC. Textbook Of Traumatic Brain Injury. Washington, DC: American Psychiatric Association. p. 34. ISBN 1-58562-105-6. Retrieved 2008-06-10.
- ↑ Granacher RP (2007). Traumatic Brain Injury: Methods for Clinical & Forensic Neuropsychiatric Assessment, Second Edition. Boca Raton: CRC. pp. 26–32. ISBN 0-8493-8138-X. Retrieved 2008-07-06.
- ↑ "The story of Roberto Guerrero".
- ↑ "Jules Bianchi: Family confirms Formula One driver sustained traumatic brain injury in Japanese GP crash". Retrieved 8 October 2014.
- ↑ "F1 driver Jules Bianchi dies from crash injuries". BBC. Retrieved 18 July 2015.
External links
- Diffuse Axonal Injury MRI and CT Images