Stability constants of complexes
A stability constant (formation constant, binding constant) is an equilibrium constant for the formation of a complex in solution. It is a measure of the strength of the interaction between the reagents that come together to form the complex. There are two main kinds of complex: compounds formed by the interaction of a metal ion with a ligand and supramolecular complexes, such as host-guest complexes and complexes of anions. The stability constant(s) provide the information required to calculate the concentration(s) of the complex(es) in solution. There are many areas of application in chemistry, biology and medicine.
History
Jannik Bjerrum developed the first general method for the determination of stability constants of metal-ammine complexes in 1941.[1] The reasons why this occurred at such a late date, nearly 50 years after Alfred Werner had proposed the correct structures for coordination complexes, have been summarised by Beck and Nagypál.[2] The key to Bjerrum's method was the use of the then recently developed glass electrode and pH meter to determine the concentration of hydrogen ions in solution. Bjerrum recognised that the formation of a metal complex with a ligand was a kind of acid–base equilibrium: there is competition for the ligand, L, between the metal ion, Mn+, and the hydrogen ion, H+. This means that there are two simultaneous equilibria that have to be considered. In what follows electrical charges are omitted for the sake of generality. The two equilibria are
- H + L ⇌ HL
- M + L ⇌ ML
Hence by following the hydrogen ion concentration during a titration of a mixture of M and HL with base, and knowing the acid dissociation constant of HL, the stability constant for the formation of ML could be determined. Bjerrum went on to determine the stability constants for systems in which many complexes may be formed.
- M + q L ⇌ MLq
The following twenty years saw a veritable explosion in the number of stability constants that were determined. Relationships, such as the Irving-Williams series were discovered. The calculations were done by hand using the so-called graphical methods. The mathematics underlying the methods used in this period are summarised by Rossotti and Rossotti.[3] The next key development was the use of a computer program, LETAGROP[4][5] to do the calculations. This permitted the examination of systems too complicated to be evaluated by means of hand-calculations. Subsequently computer programs capable of handling complex equilibria in general, such as SCOGS[6] and MINIQUAD[7] were developed so that today the determination of stability constants has almost become a "routine" operation. Values of thousands of stability constants can be found in two commercial databases.[8][9]
Theory
The formation of a complex between a metal ion, M, and a ligand, L, is in fact usually a substitution reaction. For example, in aqueous solutions, metal ions will be present as aquo ions, so the reaction for the formation of the first complex could be written as
- [M(H2O)n] + L ⇌ [M(H2O)n−1L] + H2O
The equilibrium constant for this reaction is given by
![{\displaystyle \beta ^{'}={\frac {[\mathrm {M(H_{2}O)} _{n-1}\mathrm {L} ][\mathrm {H_{2}O} ]}{[\mathrm {M(H_{2}O)} _{n}][\mathrm {L} ]}}}](../I/m/cb903e0c6849446ba718798c09f77751851f266c.svg)
[L] should be read as "the concentration of L" and likewise for the other terms in square brackets. The expression can be greatly simplified by removing those terms which are constant. The number of water molecules attached to each metal ion is constant. In dilute solutions the concentration of water is effectively constant. The expression becomes
![\beta ={\mathrm {{\frac {[ML]}{[M][L]}}}}.](../I/m/f6dbc7aecca141447456d84685c3544a3abf8308.svg)
Following this simplification a general definition can be given, For the general equilibrium
- p M + q L … ⇌ MpLq…
![{\displaystyle \beta _{pq...}={\frac {[\mathrm {M} _{p}\mathrm {L} _{q}...]}{[\mathrm {M} ]^{p}[\mathrm {L} ]^{q}...}}}](../I/m/c9b5cb845f8efec1e7800490f0c4d0e5cd90a76e.svg)
The definition can easily be extended to include any number of reagents. The reagents need not always be a metal and a ligand but can be any species which form a complex. Stability constants defined in this way, are association constants. This can lead to some confusion as pKa values are dissociation constants. In general purpose computer programs it is customary to define all constants as association constants. The relationship between the two types of constant is given in association and dissociation constants.
Stepwise and cumulative constants
A cumulative or overall constant, given the symbol β, is the constant for the formation of a complex from reagents. For example, the cumulative constant for the formation of ML2 is given by
- ;

![{\displaystyle \beta _{12}=\mathrm {\frac {[ML_{2}]}{[M][L]^{2}}} }](../I/m/f7452937a667828083cc1ec4d7c38786d67b5df9.svg)
The stepwise constants, K1 and K2 refer to the formation of the complexes one step at a time.
![{\displaystyle {\begin{array}{rl}{\ce {{M}+L<=>ML}};&K_{1}=\mathrm {\frac {[ML]}{[M][L]}} \\{\ce {{ML}+L<=>ML2}};&K_{2}=\mathrm {\frac {[ML_{2}]}{[ML][L]}} \end{array}}}](../I/m/ec1e0820c6ab351969b4c4ac7e941133313c3bd2.svg)
It follows that

A cumulative constant can always be expressed as the product of stepwise constants. Conversely, any stepwise constant can be expressed as a quotient of two or more overall constants. There is no agreed notation for stepwise constants, though a symbol such as KL
ML is sometimes found in the literature. It is best always to define each stability constant by reference to an equilibrium expression.
Hydrolysis products
The formation of a hydroxo complex is a typical example of an hydrolysis reaction. An hydrolysis reaction is one in which a substrate reacts with water, splitting a water molecule into hydroxide and hydrogen ions. In this case the hydroxide ion then forms a complex with the substrate.
- M + OH ⇌ M(OH);
![K={\mathrm {{\frac {[M(OH)]}{[M][OH]}}}}](../I/m/33c23271b9152d2329b562befa69300b63a0124c.svg)
In water the concentration of hydroxide is related to the concentration of hydrogen ions by the self-ionization constant, Kw.
- Kw=[H+][OH−]; [OH−] = Kw[H+]−1
The expression for hydroxide concentration is substituted into the formation constant expression
![{\displaystyle K={\frac {[\mathrm {M} (\mathrm {OH} )]}{[\mathrm {M} ]K_{\mathrm {w} }[\mathrm {H} ]^{-1}}}}](../I/m/15c00635c0c40ce1c98b65ad70c173b71abeedf3.svg)
![{\displaystyle \beta _{1-1}^{*}={\frac {K}{K_{\mathrm {w} }}}={\frac {[\mathrm {M} (\mathrm {OH} )]}{[\mathrm {M} ][\mathrm {H} ]^{-1}}}}](../I/m/ae990a759502a3efc0b0468f6d3122dff0f1093e.svg)
The literature usually gives value of β*.
Acid–base complexes
A Lewis acid, A, and a Lewis base, B, can be considered to form a complex AB
- A + B ⇌ AB
![{\displaystyle K=\mathrm {\frac {[AB]}{[A][B]}} }](../I/m/d802796dbfc74cdb4c54835288b81fe46b73c71e.svg)
There are three major theories relating to the strength of Lewis acids and bases and the interactions between them.
- Hard and soft acid–base theory (HSAB).[10] This is used mainly for qualitative purposes.
- Drago and Wayland proposed a two-parameter equation which predicts the standard enthalpy of formation of a very large number of adducts quite accurately. −ΔH⊖ (A − B) = EAEB + CACB. Values of the E and C parameters are available[11]
- Guttmann donor numbers: for bases the number is derived from the enthalpy of reaction of the base with antimony pentachloride in 1,2-Dichloroethane as solvent. For acids, an acceptor number is derived from the enthalpy of reaction of the acid with triphenylphosphine oxide.[12]
For more details see: acid–base reaction, acid catalysis, acid–base extraction.
Thermodynamics
The thermodynamics of metal ion complex formation provides much significant information.[13] In particular it is useful in distinguishing between enthalpic and entropic effects. Enthalpic effects depend on bond strengths and entropic effects have to do with changes in the order/disorder of the solution as a whole. The chelate effect, below, is best explained in terms of thermodynamics.
An equilibrium constant is related to the standard Gibbs free energy change for the reaction
- ΔG⊖ = −2.303 RT log10 β.
R is the gas constant and T is the absolute temperature. At 25 °C, ΔG⊖ = (−5.708 kJ mol−1) ⋅ log β. Free energy is made up of an enthalpy term and an entropy term.
- ΔG⊖ = ΔH⊖ − TΔS⊖
The standard enthalpy change can be determined by calorimetry or by using the Van 't Hoff equation, though the calorimetric method is preferable. When both the standard enthalpy change and stability constant have been determined, the standard entropy change is easily calculated from the equation above.
The fact that stepwise formation constants of complexes of the type MLn decrease in magnitude as n increases may be partly explained in terms of the entropy factor. Take the case of the formation of octahedral complexes.
- [M(H2O)mLn−1] + L ⇌ [M(H2O)m−1Ln]
For the first step m = 6, n = 1 and the ligand can go into one of 6 sites. For the second step m = 5 and the second ligand can go into one of only 5 sites. This means that there is more randomness in the first step than the second one; ΔS⊖ is more positive, so ΔG⊖ is more negative and log K1 > log K2. The ratio of the stepwise stability constants can be calculated on this basis, but experimental ratios are not exactly the same because ΔH⊖ is not necessarily the same for each step.[14] The entropy factor is also important in the chelate effect, below.
Ionic strength dependence
The thermodynamic equilibrium constant, K⊖, for the equilibrium
- M + L ⇌ ML
can be defined[15] as

where {ML} is the activity of the chemical species ML etc. K⊖ is dimensionless since activity is dimensionless. Activities of the products are placed in the numerator, activities of the reactants are placed in the denominator. See activity coefficient for a derivation of this expression.
Since activity is the product of concentration and activity coefficient (γ) the definition could also be written as
![{\displaystyle K^{\ominus }=\mathrm {\frac {[ML]}{[M][L]}} \times {\frac {\gamma _{\mathrm {ML} }}{\gamma _{\mathrm {M} }\gamma _{\mathrm {L} }}}=\mathrm {\frac {[ML]}{[M][L]}} \times \Gamma }](../I/m/73bd77587e27db8a47d8bf7dd244d05f84917022.svg)
where [ML] represents the concentration of ML and Γ is a quotient of activity coefficients. This expression can be generalized as
![{\displaystyle \beta _{pq...}^{\ominus }={\frac {[\mathrm {M} _{p}\mathrm {L} _{q}...]}{[\mathrm {M} ]^{p}[\mathrm {L} ]^{q}...}}\times \Gamma }](../I/m/07fcbbe22a4e6a23444c531c7ae018239b2f481d.svg)
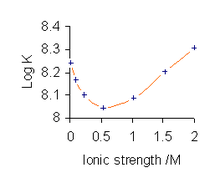
To avoid the complications involved in using activities, stability constants are determined, where possible, in a medium consisting of a solution of a background electrolyte at high ionic strength, that is, under conditions in which Γ can be assumed to be always constant.[15] For example, the medium might be a solution of 0.1 mol dm−3 sodium nitrate or 3 mol dm−3 sodium perchlorate. When Γ is constant it may be ignored and the general expression in theory, above, is obtained.
All published stability constant values refer to the specific ionic medium used in their determination and different values are obtained with different conditions, as illustrated for the complex CuL (L = glycinate). Furthermore, stability constant values depend on the specific electrolyte used as the value of Γ is different for different electrolytes, even at the same ionic strength. There does not need to be any chemical interaction between the species in equilibrium and the background electrolyte, but such interactions might occur in particular cases. For example, phosphates form weak complexes with alkali metals, so, when determining stability constants involving phosphates, such as ATP, the background electrolyte used will be, for example, a tetralkylammonium salt. Another example involves iron(III) which forms weak complexes with halide and other anions, but not with perchlorate ions.
When published constants refer to an ionic strength other than the one required for a particular application, they may be adjusted by means of specific ion theory (SIT) and other theories.[17]
Temperature dependence
All equilibrium constants vary with temperature according to the Van 't Hoff equation[18]

R is the gas constant and T is the thermodynamic temperature. Thus, for exothermic reactions, (the standard enthalpy change, ΔH⊖, is negative) K decreases with temperature, but for endothermic reactions (ΔH⊖ is positive) K increases with temperature.
Factors affecting the stability constants of complexes
The chelate effect
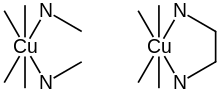
Consider the two equilibria, in aqueous solution, between the copper(II) ion, Cu2+ and ethylenediamine (en) on the one hand and methylamine, MeNH2 on the other.
-
Cu2+ + en ⇌ [Cu(en)]2+
(1)
-
Cu2+ + 2 MeNH2 ⇌ [Cu(MeNH2)2]2+
(2)
In (1) the bidentate ligand ethylene diamine forms a chelate complex with the copper ion. Chelation results in the formation of a five-membered ring. In (2) the bidentate ligand is replaced by two monodentate methylamine ligands of approximately the same donor power, meaning that the enthalpy of formation of Cu–N bonds is approximately the same in the two reactions. Under conditions of equal copper concentrations and when then concentration of methylamine is twice the concentration of ethylenediamine, the concentration of the complex (1) will be greater than the concentration of the complex (2). The effect increases with the number of chelate rings so the concentration of the EDTA complex, which has six chelate rings, is much higher than a corresponding complex with two monodentate nitrogen donor ligands and four monodentate carboxylate ligands. Thus, the phenomenon of the chelate effect is a firmly established empirical fact: under comparable conditions, the concentration of a chelate complex will be higher than the concentration of an analogous complex with monodentate ligands.
The thermodynamic approach to explaining the chelate effect considers the equilibrium constant for the reaction: the larger the equilibrium constant, the higher the concentration of the complex.
-
[Cu(en)] = β11[Cu][en]
(3)
-
[Cu(MeNH2)2] = β12[Cu][MeNH2]2
(4)
When the analytical concentration of methylamine is twice that of ethylenediamine and the concentration of copper is the same in both reactions, the concentration [Cu(en)]2+ is much higher than the concentration [Cu(MeNH2)2]2+ because β11 ≫ β12.
The difference between the two stability constants is mainly due to the difference in the standard entropy change, ΔS⊖. In equation (1) there are two particles on the left and one on the right, whereas in equation (2) there are three particles on the left and one on the right. This means that less entropy of disorder is lost when the chelate complex is formed than when the complex with monodentate ligands is formed. This is one of the factors contributing to the entropy difference. Other factors include solvation changes and ring formation. Some experimental data to illustrate the effect are shown in the following table.[19]
Equilibrium log β ΔG⊖ ΔH⊖ /kJ mol−1 −TΔS⊖ /kJ mol−1 Cd2+ + 4 MeNH2 ⇌ Cd(MeNH
2)2+
46.55 −37.4 −57.3 19.9 Cd2+ + 2 en ⇌ Cd(en)2+
210.62 −60.67 −56.48 −4.19
These data show that the standard enthalpy changes are indeed approximately equal for the two reactions and that the main reason why the chelate complex is so much more stable is that the standard entropy term is much less unfavourable, indeed, it is favourable in this instance. In general it is difficult to account precisely for thermodynamic values in terms of changes in solution at the molecular level, but it is clear that the chelate effect is predominantly an effect of entropy. Other explanations, including that of Schwarzenbach,[20] are discussed in Greenwood and Earnshaw.[19]
The chelate effect increases as the number of chelate rings increases. For example, the complex [Ni(dien)2)]2+ is more stable than the complex [Ni(en)3)]2+; both complexes are octahedral with six nitrogen atoms around the nickel ion, but dien (diethylenetriamine, 1,4,7-triazaheptane) is a tridentate ligand and en is bidentate. The number of chelate rings is one less than the number of donor atoms in the ligand. EDTA (ethylenediaminetetracetic acid) has six donor atoms so it forms very strong complexes with five chelate rings. Ligands such as DTPA, which have eight donor atoms are used to form complexes with large metal ions such as lanthanide or actinide ions which usually form 8- or 9-coordinate complexes.
5-membered and 6-membered chelate rings give the most stable complexes. 4-membered rings are subject to internal strain because of the small inter-bond angle is the ring. The chelate effect is also reduced with 7- and 8- membered rings, because the larger rings are less rigid, so less entropy is lost in forming them.
Deprotonation of aliphatic –OH groups

Removal of a proton from an aliphatic –OH group is difficult to achieve in aqueous solution because the energy required for this process is rather large. Thus, ionization of aliphatic –OH groups occurs in aqueous solution only in special circumstances. One such circumstance is found with compounds containing the H2N–C–C–OH substructure. For example, compounds containing the aminoethanol substructure can form metal–chelate complexes with the deprotonated form, H2N–C–C–O−. The chelate effect supplies the extra energy needed to break the –OH bond.
An important example occurs with the molecule tris. This molecule should be used with caution as a buffering agent as it will form chelate complexes with ions such as Fe3+ and Cu2+.
The macrocyclic effect
It was found that the stability of the complex of copper(II) with the macrocyclic ligand cyclam (1,4,8,11-tetraazacyclotetradecane) was much greater than expected in comparison to the stability of the complex with the corresponding open-chain amine.[21] This phenomenon was named "the macrocyclic effect" and it was also interpreted as an entropy effect. However, later studies suggested that both enthalpy and entropy factors were involved.[22]
An important difference between macrocyclic ligands and open-chain (chelating) ligands is that they have selectivity for metal ions, based on the size of the cavity into which the metal ion is inserted when a complex is formed. For example, the crown ether 18-crown-6 forms much stronger complexes with the potassium ion, K+ than with the smaller sodium ion, Na+.[23]
In hemoglobin an iron(II) ion is complexed by a macrocyclic porphyrin ring. The article hemoglobin incorrectly states that oxyhemoglogin contains iron(III). It is now known that the iron(II) in hemoglobin is a low-spin complex, whereas in oxyhemoglobin it is a high-spin complex. The low-spin Fe2+ ion fits snugly into the cavity of the porhyrin ring, but high-spin iron(II) is significantly larger and the iron atom is forced out of the plane of the macrocyclic ligand.[24] This effect contributes the ability of hemoglobin to bind oxygen reversibly under biological conditions. In Vitamin B12 a cobalt(II) ion is held in a corrin ring. Chlorophyll is a macrocyclic complex of magnesium(II).
 |
 |
| Cyclam | Porphine, the simplest porphyrin. |
.png) |
| Structures of common crown ethers: 12-crown-4, 15-crown-5, 18-crown-6, dibenzo-18-crown-6, and diaza-18-crown-6 |
Geometrical factors
Successive stepwise formation constants Kn in a series such as MLn (n = 1, 2, ...) usually decrease as n increases. Exceptions to this rule occur when the geometry of the MLn complexes is not the same for all members of the series. The classic example is the formation of the diamminesilver(I) complex [Ag(NH3)2]+ in aqueous solution.
![{\displaystyle {\begin{array}{rl}{\ce {{Ag+}+NH3<=>{[Ag(NH3)]+}}}&K_{1}={\frac {\left[{\ce {[Ag(NH3)]+}}\right]}{\ce {[Ag+][NH3]}}}\\{\ce {{Ag(NH3)+}+NH3<=>{[Ag(NH3)2]+}}}&K_{2}={\frac {\left[{\ce {[Ag(NH3)2]+}}\right]}{\ce {[[Ag(NH3)]+][NH3]}}}\end{array}}}](../I/m/df098fa2168f16644e51719486b5fcaf5b50306c.svg)
In this case, K2 > K1. The reason for this is that, in aqueous solution, the ion written as Ag+ actually exists as the four-coordinate tetrahedral aqua species [Ag(OH2)4]+. The first step is then a substitution reaction involving the displacement of a bound water molecule by ammonia forming the tetrahedral complex [Ag(NH3)(OH2)3]+ (commonly abbreviated as [Ag(NH3)]+). In the second step, the aqua ligands are lost to form a linear, two-coordinate product [H3N–Ag–NH3]+. Examination of the thermodynamic data shows that both enthalpy and entropy effects determine the result.[25]
| equilibrium | ΔH⊖ /kJ mol−1 | ΔS⊖ /J K−1 mol−1 |
|---|---|---|
| Ag+ + NH3 ⇌ [Ag(NH3)]+ | −21.4 | 8.66 |
| [Ag(NH3)]+ + NH3 ⇌ [Ag(NH3)2]+ | −35.2 | −61.26 |
Other examples exist where the change is from octahedral to tetrahedral, as in the formation of [CoCl4]2− from [Co(H2O)6]2+.
Classification of metal ions
Ahrland, Chatt and Davies proposed that metal ions could be described as class A if they formed stronger complexes with ligands whose donor atoms are nitrogen, oxygen or fluorine than with ligands whose donor atoms are phosphorus, sulfur or chlorine and class B if the reverse is true.[26] For example, Ni2+ forms stronger complexes with amines than with phosphines, but Pd2+ forms stronger complexes with phosphines than with amines. Later, Pearson proposed the theory of hard and soft acids and bases (HSAB theory).[27] In this classification, class A metals are hard acids and class B metals are soft acids. Some ions, such as copper(I), are classed as borderline. Hard acids form stronger complexes with hard bases than with soft bases. In general terms hard–hard interactions are predominantly electrostatic in nature whereas soft–soft interactions are predominantly covalent in nature. The HSAB theory, though useful, is only semi-quantitative.[28]
The hardness of a metal ion increases with oxidation state. An example of this effect is given by the fact that Fe2+ tends to form stronger complexes with N-donor ligands than with O-donor ligands, but the opposite is true for Fe3+.
Effect of ionic radius
The Irving–Williams series refers to high-spin, octahedral, divalent metal ion of the first transition series. It places the stabilities of complexes in the order
- Mn < Fe < Co < Ni < Cu > Zn
This order was found to hold for a wide variety of ligands.[29] There are three strands to the explanation of the series.
- The ionic radius is expected to decrease regularly for Mn2+ to Zn2+. This would be the normal periodic trend and would account for the general increase in stability.
- The crystal field stabilisation energy (CFSE) increases from zero for manganese(II) to a maximum at nickel(II). This makes the complexes increasingly stable. CFSE returns to zero for zinc(II).
- Although the CFSE for copper(II) is less than for nickel(II), octahedral copper(II) complexes are subject to the Jahn–Teller effect which results in a complex having extra stability.
Another example of the effect of ionic radius the steady increase in stability of complexes with a given ligand along the series of trivalent lanthanide ions, an effect of the well-known lanthanide contraction.
Applications
Stability constant values are exploited in a wide variety of applications. Chelation therapy is used in the treatment of various metal-related illnesses, such as iron overload in β-thalassemia sufferers who have been given blood transfusions. The ideal ligand binds to the target metal ion and not to others, but this degree of selectivity is very hard to achieve. The synthetic drug deferiprone achieves selectivity by having two oxygen donor atoms so that it binds to Fe3+ in preference to any of the other divalent ions that are present in the human body, such as Mg2+, Ca2+ and Zn2+. Treatment of poisoning by ions such as Pb2+ and Cd2+ is much more difficult since these are both divalent ions and selectivity is harder to accomplish.[30] Excess copper in Wilson's disease can be removed by penicillamine or Triethylene tetramine (TETA). DTPA has been approved by the U.S. Food and Drug Administration for treatment of plutonium poisoning.
DTPA is also used as a complexing agent for gadolinium in MRI contrast enhancement. The requirement in this case is that the complex be very strong, as Gd3+ is very toxic. The large stability constant of the octadentate ligand ensures that the concentration of free Gd3+ is almost negligible, certainly well below toxicity threshold.[31] In addition the ligand occupies only 8 of the 9 coordination sites on the gadolinium ion. The ninth site is occupied by a water molecule which exchanges rapidly with the fluid surrounding it and it is this mechanism that makes the paramagnetic complex into a contrast reagent.
EDTA forms such strong complexes with most divalent cations that it finds many uses. For example, it is often present in washing powder to act as a water softener by sequestering calcium and magnesium ions.
The selectivity of macrocyclic ligands can be used as a basis for the construction of an ion selective electrode. For example, potassium selective electrodes are available that make use of the naturally-occurring macrocyclic antibiotic valinomycin.
 |
 |
 | |
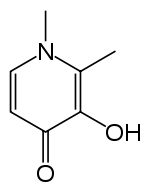
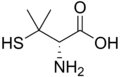
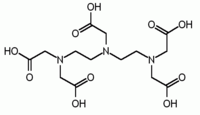
| Deferiprone | Penicillamine | triethylenetetramine, TETA | Ethylenediaminetetraacetic acid, EDTA |
 |
 |
 | |
| Diethylenetriaminepentaacetic acid, DTPA | Valinomycin | Tri-n-butyl phosphate |
An ion-exchange resin such as chelex 100, which contains chelating ligands bound to a polymer, can be used in water softeners and in chromatographic separation techniques. In solvent extraction the formation of electrically-neutral complexes allows cations to be extracted into organic solvents. For example, in nuclear fuel reprocessing uranium(VI) and plutonium(VI) are extracted into kerosene as the complexes [MO2(TBP)2(NO3)2] (TBP = tri-n-butyl phosphate). In phase-transfer catalysis, a substance which is insoluble in an organic solvent can be made soluble by addition of a suitable ligand. For example, potassium permanganate oxidations can be achieved by adding a catalytic quantity of a crown ether and a small amount of organic solvent to the aqueous reaction mixture, so that the oxidation reaction occurs in the organic phase.
In all these examples, the ligand is chosen on the basis of the stability constants of the complexes formed. For example, TBP is used in nuclear fuel reprocessing because (among other reasons) it forms a complex strong enough for solvent extraction to take place, but weak enough that the complex can be destroyed by nitric acid to recover the uranyl cation as nitrato complexes, such as [UO2(NO3)4]2− back in the aqueous phase.
Supramolecular complexes
Supramolecular complexes are held together by hydrogen bonding, hydrophobic forces, van der Waals forces, π-π interactions, and electrostatic effects, all of which can be described as noncovalent bonding. Applications include molecular recognition, host-guest chemistry and anion sensors.
A typical application in molecular recognition involved the determination of formation constants for complexes formed between a tripodal substituted urea molecule and various saccharides.[32] The study was carried out using a non-aqueous solvent and NMR chemical shift measurements. The object was to examine the selectivity with respect to the saccharides.
An example of the use of supramolecular complexes in the development of chemosensors is provided by the use of transition-metal ensembles to sense for ATP.[33]
Anion complexation can be achieved by encapsulating the anion in a suitable cage. Selectivity can be engineered by designing the shape of the cage. For example, dicarboxylate anions could be encapsulated in the ellipsoidal cavity in a large macrocyclic structure containing two metal ions.[34]
Experimental methods
The method developed by Bjerrum is still the main method in use today, though the precision of the measurements has greatly increased. Most commonly, a solution containing the metal ion and the ligand in a medium of high ionic strength is first acidified to the point where the ligand is fully protonated. This solution is then titrated, often by means of a computer-controlled auto-titrator, with a solution of CO2-free base. The concentration, or activity, of the hydrogen ion is monitored by means of a glass electrode. The data set used for the calculation has three components: a statement defining the nature of the chemical species that will be present, called the model of the system, details concerning the concentrations of the reagents used in the titration, and finally the experimental measurements in the form of titre and pH (or emf) pairs.
It is not always possible to use a glass electrode. If that is the case, the titration can be monitored by other types of measurement. Absorbance spectra, fluorescence spectra and NMR spectra are the most commonly used alternatives. Current practice is to take absorbance or fluorescence measurements at a range of wavelengths and to fit these data simultaneously. Various NMR chemical shifts can also be fitted together.
The chemical model will include values of the protonation constants of the ligand, which will have been determined in separate experiments, a value for log Kw and estimates of the unknown stability constants of the complexes formed. These estimates are necessary because the calculation uses a non-linear least-squares algorithm. The estimates are usually obtained by reference to a chemically similar system. The stability constant databases[8][9] can be very useful in finding published stability constant values for related complexes.
In some simple cases the calculations can be done in a spreadsheet.[35] Otherwise, the calculations are performed with the aid of a general-purpose computer programs. The most frequently used programs are:
- Potentiometric and/or spectrophotometric data: PSEQUAD[36]
- Potentiometric data: HYPERQUAD,[37] BEST[38]
- Spectrophotometric data: HypSpec, SQUAD,[39] SPECFIT,[40][41] ReactLab EQUILIBRIA.[42]
- NMR data HypNMR,[43] WINEQNMR2[44]
In biochemistry, formation constants of adducts may be obtained from Isothermal titration calorimetry (ITC) measurements. This technique yields both the stability constant and the standard enthalpy change for the equilibrium.[45] It is mostly limited, by availability of software, to complexes of 1:1 stoichiometry.
Critically evaluated data
The following references are for critical reviews of published stability constants for various classes of ligands. All these reviews are published by IUPAC and the full text is available, free of charge, in pdf format.
- ethylenediamine (en) [46]
- Nitrilotriacetic acid (NTA)[47]
- aminopolycarboxylic acids (complexones)[48]
- Alpha hydroxy acids and other hydroxycarboxylic acids[49][50]
- amino acids with polar side-chains[55]
- general[58]
- Chemical speciation of environmentally significant heavy metals with inorganic ligands. Part 1: The Hg2+–Cl−, OH−, CO2−
3, SO2−
4, and PO3−
4 systems.[59] - Chemical speciation of environmentally significant metals with inorganic ligands Part 2: The Cu2+–OH−, Cl−, CO2−
3, SO2−
4, and PO3−
4 aqueous systems[60] - Chemical speciation of environmentally significant metals with inorganic ligands Part 3: The Pb2+–OH−, Cl−, CO2−
3, SO2−
4, and PO3−
4 systems[61] - Chemical speciation of environmentally significant metals with inorganic ligands. Part 4: The Cd2+–OH−, Cl−, CO2−
3, SO2−
4, and PO3−
4 systems[62]
Databases
- The Ki Database is a public domain database of published binding affinities (Ki) of drugs and chemical compounds for receptors, neurotransmitter transporters, ion channels, and enzymes.
- BindingDB is a public domain database of measured binding affinities, focusing chiefly on the interactions of protein considered to be drug-targets with small, drug-like molecules
References
- ↑ Bjerrum, J. (1941). Metal-ammine formation in aqueous solution. Copenhagen: Haase.
- ↑ Beck, M. T.; Nagypál, I. (1990). "Chapter 1". Chemistry of Complex Equilibria. Horwood. ISBN 0-85312-143-5.
- ↑ Rossotti, F. J. C.; Rossotti, H. (1961). The Determination of Stability Constants. McGraw–Hill.
- ↑ Dyrssen, D.; Ingri, N.; Sillen, L. G. (1961). "Pit-mapping - A general approach to Computer refinement of stability constants.". Acta Chem. Scand. 15: 694–696. doi:10.3891/acta.chem.scand.15-0694.
- ↑ Ingri, N.; Sillen, L. G. (1964). "High-speed computers as a supplement to graphical methods, IV. An ALGOL version of LETAGROP-VRID". Arkiv för Kemi. 23: 97–121.
- ↑ Sayce, I. G. (1968). "Computer calculations of equilibrium constantsof species present in mixtures of metal ions and complexing reagents". Talanta. 22 (12): 1397–1421. doi:10.1016/0039-9140(68)80200-0. PMID 18960446.
- ↑ Sabatini, A.; Vacca, A.; Gans, P. (1974). "MINIQUAD - A general computer program for the computation of Stability constants". Talanta. 21 (1): 53–77. doi:10.1016/0039-9140(74)80063-9. PMID 18961420.
- 1 2 IUPAC SC-Database A comprehensive database of published data on equilibrium constants of metal complexes and ligands
- 1 2 NIST Standard Reference Database 46 Critically Selected Stability Constants of Metal Complexes
- ↑ Pearson, R. G. (1997). Chemical Hardness: Applications from Molecules to Solids. Springer-VCH. ISBN 3-527-29482-1.
- ↑ Drago, R. S.; Wong, N.; Bilgrien, C.; Vogel, C. (1987). "E and C parameters from Hammett substituent constants and use of E and C to understand cobalt–carbon bond energies". Inorg. Chem. 26 (1): 9–14. doi:10.1021/ic00248a003.
- ↑ Gutmann, V. (1978). The Donor–Acceptor Approach to Molecular Interactions. Springer. ISBN 0-306-31064-3.
- ↑ Rossotti, F. J. C. (1960). "The thermodynamics of metal ion complex formation in solution". In Lewis, J.; R. G., Wilkins. Modern coordination chemistry. New York: Interscience.
- ↑ Beck, M. T.; Nagypál, I. (1990). Chemistry of Complex Equilibria. Horwood. ISBN 0-85312-143-5. sections 3.5.1.2, 6.6.1 and 6.6.2
- 1 2 Rossotti, F. J. C.; Rossotti, H. (1961). "Chapter 2: Activity and Concentration Quotients". The Determination of Stability Constants. McGraw–Hill.
- ↑ Gergely, A.; Nagypál, I.; E., Farkas (1974). "A réz(II)-aminosav törzskomplexek vizes oldatában lejátszodó protoncsere-reakciók kinetikájának NMR-vizsgálata" [NMR study of the proton exchange process in aqueous solutions of copper(II)-aminoacid parent complexes]. Magyar Kémiai Folyóirat. 80: 545–549.
- ↑ "Project: Ionic Strength Corrections for Stability Constants". IUPAC. Retrieved 2008-11-23.
- ↑ Atkins, P. W.; De Paula, J. (2006). "Section 7.4: The Response of Equilibria to Temperature". Physical Chemistry. Oxford University Press. ISBN 0-19-870072-5.
- 1 2 Greenwood, Norman N.; Earnshaw, Alan (1997). Chemistry of the Elements (2nd ed.). Butterworth-Heinemann. ISBN 0-08-037941-9. p. 910
- ↑ Schwarzenbach, G. (1952). "Der Chelateffekt" [The Chelate Effect]. Helv. Chim. Acta. 35 (7): 2344–2359. doi:10.1002/hlca.19520350721.
- ↑ Cabinness, D. K.; Margerum, D. W. (1969). "Macrocyclic effect on the stability of copper(II) tetramine complexes". J. Am. Chem. Soc. 91 (23): 6540–6541. doi:10.1021/ja01051a091.
- ↑ Lindoy, L. F. (1990). "Chapter 6: Thermodynamic considerations". The Chemistry of Macrocyclic Ligand Complexes. Cambridge University Press. ISBN 0-521-40985-3.
- ↑ Pedersen, C. J. (1967). "Cyclic polyethers and their complexes with metal salts". J. Am. Chem. Soc. 89 (26): 7017–7036. doi:10.1021/ja01002a035.
- ↑ Greenwood, Norman N.; Earnshaw, Alan (1997). Chemistry of the Elements (2nd ed.). Butterworth-Heinemann. ISBN 0-08-037941-9. p. 1100, Figure 25.7
- ↑ Lundeen, M.; Hugus, Z. Z. (1992). "A calorimetric study of some metal ion complexing equilibria". Thermochim. Acta. 196 (1): 93–103. doi:10.1016/0040-6031(92)85009-K.
- ↑ Ahrland, S.; Chatt, J.; Davies, N. R. (1958). "The relative affinities of ligand atoms for acceptor molecules and ions". Quart. Rev. 12 (3): 265–276. doi:10.1039/QR9581200265.
- ↑ Pearson, R. G. (1963). "Hard and Soft Acids and Bases". J. Am. Chem. Soc. 85 (22): 3533–3539. doi:10.1021/ja00905a001.
- ↑ Beck, M. T.; Nagypál, I. (1990). Chemistry of Complex Equilibria. Horwood. p. 354. ISBN 0-85312-143-5.
- ↑ Irving, H. M. N. H.; Williams, R. J. P. (1953). "The stability of transition-metal complexes". J. Chem. Soc.: 3192–3210. doi:10.1039/JR9530003192.
- ↑ Arena, G.; Contino, A.; Longo, E.; Sciotto, D.; Spoto, G. (2001). "Selective complexation of soft Pb2+ and Hg2+ by a novel allyl functionalized thioamide calix[4]arene in 1,3-alternate conformation: a UV-visible and H-1 NMR spectroscopic investigation". J. Chem. Soc., Perkin Trans. 2 (12): 2287–2291. doi:10.1039/b107025h.
- ↑ Runge, V. M.; Scott, S. (1998). Contrast-enhanced Clinical Magnetic Resonance Imaging. University Press of Kentucky. ISBN 0-8131-1944-8.
- ↑ Vacca, A.; Nativi, C.; Cacciarini, M.; Pergoli, R.; Roelens, S. (2004). "A New Tripodal Receptor for Molecular Recognition of Monosaccharides. A Paradigm for Assessing Glycoside Binding Affinities and Selectivities by 1H NMR Spectroscopy". J. Am. Chem. Soc. 126 (50): 16456–16465. doi:10.1021/ja045813s. PMID 15600348.
- ↑ Marcotte, N.; Taglietti, A. (2003). "Transition-metal-based chemosensing ensembles: ATP sensing in physiological conditions". Supramol. Chem. 15 (7): 617–717. doi:10.1080/10610270310001605205.
- ↑ Boiocchi, M.; Bonizzoni, M.; Fabbrizzi, L.; Piovani, G.; Taglietti, A. (2004). "A dimetallic cage with a long ellipsoidal cavity for the fluorescent detection of dicarboxylate anions in water". Angew. Chem. Int. Ed. 43 (29): 3847–3852. doi:10.1002/anie.200460036. PMID 15258953.
- ↑ Billo, E. J. (1997). "Chapter 22". Excel for Chemists: A comprehensive guide (2nd ed.). Wiley-VCH. ISBN 0-471-18896-4.
- ↑ Zekany, L.; Nagypál, I. (1985). "Chapter 8: PSEQUAD: A comprehensive program for the evaluation of potentiometric and/or spectrophotometric equilibrium data using analytical derivatives". In Leggett. Computational methods for the determination of formation constants. Plenum. ISBN 0-306-41957-2.
- ↑ Gans, P.; Sabatini, A.; Vacca, A. (1996). "Investigation of equilibria in solution. Determination of equilibrium constants with the HYPERQUAD suite of programs". Talanta. 43 (10): 1739–1753. doi:10.1016/0039-9140(96)01958-3. PMID 18966661.
- ↑ Martell, A. E.; Motekaitis, R. J. (1992). The determination and use of stability constants. Wiley-VCH.
- ↑ Leggett, D. (1985). "Chapter 6: SQUAD: Stability quotients from absorbance data". In Leggett. Computational methods for the determination of formation constants. Plenum. ISBN 0-306-41957-2.
- ↑ Gampp, M.; Maeder, M.; Mayer, C. J.; Zuberbühler, A. D. (1985). "Calculation of equilibrium constants from multiwavelength spectroscopic data—I : Mathematical considerations". Talanta. 32 (2): 95–101. doi:10.1016/0039-9140(85)80035-7. PMID 18963802.
- ↑ Gampp, M.; Maeder, M.; Mayer, C. J.; Zuberbühler, A. D. (1985). "Calculation of equilibrium constants from multiwavelength spectroscopic data—II1: Specfit: two user-friendly programs in basic and standard fortran 77". Talanta. 1995. 32 (4): 251–264. doi:10.1016/0039-9140(85)80077-1. PMID 18963840.
- ↑ Jplus Consulting Pty Ltd
- ↑ Frassineti, C.; Alderighi, L.; Gans, P.; Sabatini, A.; Vacca, A.; Ghelli, S. (2003). "Determination of protonation constants of some fluorinated polyamines by means of 13C NMR data processed by the new computer program HypNMR2000. Protonation sequence in polyamines". Anal. Bioanal. Chem. 376 (7): 1041–1052. doi:10.1007/s00216-003-2020-0. PMID 12845401.
- ↑ Hynes, M. J. (1993). "EQNMR: A computer program for the calculation of stability constants from nuclear magnetic resonance chemical shift data". J. Chem. Soc., Dalton Trans. (2): 311–312. doi:10.1039/DT9930000311.
- ↑ O'Brien, R.; Ladbury, J. E.; Chowdry, B. Z. (2000). "Chapter 10". In Harding, S. E.; Chowdry, B. Z. Protein-Ligand interactions: hydrodynamics and calorimetry. Oxford University Press. ISBN 0-19-963749-0.
- ↑ Paoletti, P. (1984). "Formation of metal complexes with ethylenediamine: a critical survey of equilibrium constants, enthalpy and entropy values" (PDF). Pure Appl. Chem. 56 (4): 491–522. doi:10.1351/pac198456040491.
- ↑ Anderegg, G. (1982). "Critical survey of stability constants of NTA complexes" (PDF). Pure Appl. Chem. 54 (12): 2693–2758. doi:10.1351/pac198254122693.
- ↑ Anderegg, G; Arnaud-Neu, F.; Delgado, R.; Felcman, J.; Popov, K. (2003). "Critical evaluation of stability constants of metal complexes of complexones for biomedical and environmental applications (IUPAC Technical Report)" (PDF). Pure Appl. Chem. 77 (8): 1445–95. doi:10.1351/pac200577081445.
- ↑ Lajunen, L. H. J.; Portanova, R.; Piispanen, J.; Tolazzi, M. (1997). "Critical evaluation of stability constants for alpha-hydroxycarboxylic acid complexes with protons and metal ions and the accompanying enthalpy changes. Part I: Aromatic ortho-hydroxycarboxylic acids (Technical Report)" (PDF). Pure Appl. Chem. 69 (2): 329–382. doi:10.1351/pac199769020329.
- ↑ Portanova, R; Lajunen, L. H. J.; Tolazzi, M.; Piispanen, J. (2003). "Critical evaluation of stability constants for alpha-hydroxycarboxylic acid complexes with protons and metal ions and the accompanying enthalpy changes. Part II. Aliphatic 2-hydroxycarboxylic acids (IUPAC Technical Report)" (PDF). Pure Appl. Chem. 75 (4): 495–540. doi:10.1351/pac200375040495.
- ↑ Arnaud-Neu, F.; Delgado, R.; Chaves, S. (2003). "Critical evaluation of stability constants and thermodynamic functions of metal complexes of crown ethers (IUPAC Technical Report)" (PDF). Pure Appl. Chem. 75 (1): 71–102. doi:10.1351/pac200375010071.
- ↑ Popov, K.; Rönkkömäki, H.; Lajunen, L. H. J. (2001). "Critical evaluation of stability constants of phosphonic acids (IUPAC Technical Report)" (PDF). Pure Appl. Chem. 73 (11): 1641–1677. doi:10.1351/pac200173101641.
- ↑ Popov, K.; Rönkkömäki, H.; Lajunen, L. H. J. (2002). "Errata" (PDF). Pure Appl. Chem. 74 (11): 2227–2227. doi:10.1351/pac200274112227.
- ↑ Sjöberg, S. (1997). "Critical evaluation of stability constants of metal-imidazole and metal-histamine systems (Technical Report)" (PDF). Pure Appl. Chem. 69 (7): 1549–1570. doi:10.1351/pac199769071549.
- ↑ Berthon, G. (1995). "Critical evaluation of the stability constants of metal complexes of amino acids with polar side chains (Technical Report)" (PDF). Pure Appl. Chem. 67 (7): 1117–1240. doi:10.1351/pac199567071117.
- ↑ Smith, R. M.; Martell, A. E.; Chen, Y. (1991). "Critical evaluation of stability constants for nucleotide complexes with protons and metal ions and the accompanying enthalpy changes" (PDF). Pure Appl. Chem. 63 (7): 1015–1080. doi:10.1351/pac199163071015.
- ↑ Stary, J.; Liljenzin, J. O. (1982). "Critical evaluation of equilibrium constants involving acetylacetone and its metal chelates" (PDF). Pure Appl. Chem. 54 (12): 2557–2592. doi:10.1351/pac198254122557.
- ↑ Beck, M. T. (1977). "Critical evaluation of equilibrium constants in solution. Stability constants of metal complexes" (PDF). Pure Appl. Chem. 49 (1): 127–136. doi:10.1351/pac197749010127.
- ↑ Powell, Kipton, J.; Brown, Paul L.; Byrne, Robert H.; Gajda, Tamás; Hefter, Glenn; Sjöberg, Staffan; Wanner, Hans (2005). "Chemical speciation of environmentally significant heavy metals with inorganic ligands. Part 1: The Hg2+–Cl−, OH−, CO2−
3, SO2−
4, and PO3−
4 aqueous systems" (PDF). Pure Appl. Chem. 77 (4): 739–800. doi:10.1351/pac200577040739. - ↑ Powell, Kipton J.; Brown, Paul L.; Byrne, Robert H.; Gajda, Tamás; Hefter, Glenn; Sjöberg, Staffan; Wanner, Hans (2007). "Chemical speciation of environmentally significant metals with inorganic ligands Part 2: The Cu2+–OH−, Cl−, CO2−
3, SO2−
4, and PO3−
4 systems" (PDF). Pure Appl. Chem. 79 (5): 895–950. doi:10.1351/pac200779050895. - ↑ Powell, Kipton J.; Brown, Paul L.; Byrne, Robert H.; Gajda, Tamás; Hefter, Glenn; Leuz, Ann-Kathrin; Sjöberg, Staffan; Wanner, Hans (2009). "Chemical speciation of environmentally significant metals with inorganic ligands Part 3: The Pb2+–OH−, Cl−, CO2−
3, SO2−
4, and PO3−
4 systems" (PDF). Pure Appl. Chem. 81 (12): 2425–2476. doi:10.1351/PAC-REP-09-03-05. - ↑ Powell, Kipton J.; Brown, Paul L.; Byrne, Robert H.; Gajda, Tamás; Hefter, Glenn; Leuz, Ann-Kathrin; Sjöberg, Staffan; Wanner, Hans (2011). "Chemical speciation of environmentally significant metals with inorganic ligands. Part 4: The Cd2+–OH−, Cl−, CO2−
3, SO2−
4, and PO3−
4 systems" (PDF). Pure Appl. Chem. 83 (5): 1163–1214. doi:10.1351/PAC-REP-10-08-09.
Further reading
Sigel, Roland K. O.; Skilandat, Miriam; Sigel, Astrid; Operschall, Bert P.; Sigel, Helmut (2013). "Chapter 8. Complex formation of cadmium with sugar residues, nucleobases, phosphates, nucleotides and nucleic acids". In Sigel, Astrid; Sigel, Helmut; Sigel, Roland K. O. Cadmium: From Toxicology to Essentiality. Metal Ions in Life Sciences. 11. Springer. pp. 191–274. doi:10.1007/978-94-007-5179-8_8.
Sóvágó, Imre; Várnagy, Katalin (2013). "Chapter 9. Cadmium(II) complexes of amino acids and peptides". In Sigel, Astrid; Sigel, Helmut; Sigel, Roland K. O. Cadmium: From Toxicology to Essentiality. Metal Ions in Life Sciences. 11. Springer. pp. 275–302. doi:10.1007/978-94-007-5179-8_9.
Yatsimirsky, Konstantin Borisovich; Vasilyev, Vladimir Pavlovich (1960). Instability Constants of Complex Compounds. Translated by Patterson, D. A. OUP.
External links
- Stability constants website: Contains information on computer programs, applications, databases and hardware for experimental titrations.