G2-M DNA damage checkpoint
The G2-M DNA damage checkpoint is an important cell cycle checkpoint in eukaryotic organisms ranging from yeast to mammals. This checkpoint ensures that cells don't initiate mitosis before they have a chance to repair damaged DNA after replication. Cells that have a defective G2-M checkpoint enter mitosis before repairing their DNA, leading to death after cell division.[1]
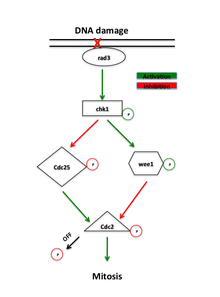
The cell cycle is driven by proteins called cyclin dependent kinases that associate with cyclin regulatory proteins at different points of the cell cycle. Accumulation of cyclin B increases the activity of the cyclin dependent kinase cdc2 as cells prepare to enter mitosis. Cdc2 activity is further regulated by phosphorylation of its tyrosine-15 residue by the kinase wee1. Phosphorylation of tyrosine-15 inhibits cdc2 activity while dephosphorylation by the phosphatase cdc25 activates the mitotic kinase.[2]
Proteins that localize to sites of DNA damage in the G2 phase initiate a signaling cascade that regulates wee1 and cdc25 activity, therefore controlling mitotic entry via cdc2-cyclin B. Delay in mitotic entry is important for cells to repair any DNA damage that may have accumulated after S phase. Absence of wee1 or removal of the tyrosine-15 site removes negative regulation of cdc2 activity and causes cells to enter mitosis without completing repair, which effectively abolishes the G2-M checkpoint.[3] Absence of cdc25 arrests cells in G2, but still allows activation of the G2-M checkpoint, implicating activation of wee1 and deactivation of cdc25 as important regulatory steps in the checkpoint.[4]
Proteins that function in the G2-M checkpoint were originally identified in yeast screens that looked for mutants which show enhanced sensitivity to radiation, termed "rad" mutants.[1] Inefficient repair of DNA damaged by ionizing radiation or chemical agents in these mutants revealed proteins essential in this pathway. Early signaling proteins in the checkpoint pathway are members of a family of phosphotidylinositol 3-kinases, rad3 in yeast and ATR in vertebrates, that are believed to localize to sites of DNA damage.[5] Rad3 phosphorylates rad26 which is required to initiate, but not maintain the checkpoint. Rad3 also phosphorylates a number of other proteins whose absence abolishes checkpoint DNA repair, including rad1, rad9, hus1 and rad17.[1] It has been hypothesized that rad9, hus 1 and rad 17 are similar to proteins involved in forming the clamp that increases the processivity of DNA polymerase during DNA replication.[6] In agreement with this idea, rad17 is similar to proteins involved in loading the clamp onto DNA. This supports a model where phosphorylation by rad3 causes recruitment of these proteins to sites of DNA damage where they mediate the activity of DNA polymerases involved in DNA repair.[1]
The main rad3 effector is the kinase chk1, which is required for the G2-M arrest in response to DNA-damaging agents.[7] This kinase is phosphorylated by rad3 between S phase and mitosis, implicating its specific role in G2 arrest. Overexpression of chk1 also rescues the radiation sensitivity of rad mutants, presumably by allowing DNA repair to take place before entry into mitosis.[5] This effect is dependent on the presence of wee1 and cdc25, which places chk1 upstream of these direct regulators of cyclin-dependent kinase. Phosphorylation activates chk1, which then phosphorylates wee1, increasing its stability.[4] Activated chk1 also phosphorylates cdc25, either leading to its inactivation or preventing its localization to the nucleus.[8] An increasingly stable wee1 further inhibits cdc2 activity, while an inactive cdc25 prevents removal of this inhibition, resulting in a strong G2 arrest.[1]
Rad3 is required for activation of chk1 and initiation of G2 arrest, but different proteins are believed to maintain G2 arrest so that DNA repair can occur. One such protein is rad18 that is required for G2 arrest even when chk1 is phosphorylated and active.[9] This protein is also involved in DNA repair, since rad18 mutants aren't able to repair DNA even when G2 arrest is prolonged by other means.
References
- 1 2 3 4 5 Cuddihy, A. R.; O'Connell, M. J. (2003). "Cell-cycle responses to DNA damage in G2". International review of cytology. 222: 99–140. doi:10.1016/s0074-7696(02)22013-6. PMID 12503848.
- ↑ Gould, K. L.; Nurse, P. (1989). "Tyrosine phosphorylation of the fission yeast cdc2+ protein kinase regulates entry into mitosis". Nature. 342 (6245): 39–45. doi:10.1038/342039a0. PMID 2682257.
- ↑ Lundgren, K.; Walworth, N.; Booher, R.; Dembski, M.; Kirschner, M.; Beach, D. (1991). "Mik1 and wee1 cooperate in the inhibitory tyrosine phosphorylation of cdc2". Cell. 64 (6): 1111–1122. doi:10.1016/0092-8674(91)90266-2. PMID 1706223.
- 1 2 Raleigh, J. M.; O'Connell, M. J. (2000). "The G(2) DNA damage checkpoint targets both Wee1 and Cdc25". Journal of Cell Science. 113 (10): 1727–1736. PMID 10769204.
- 1 2 Al-Khodairy, F.; Carr, A. M. (1992). "DNA repair mutants defining G2 checkpoint pathways in Schizosaccharomyces pombe". The EMBO Journal. 11 (4): 1343–1350. PMC 556583
 . PMID 1563350.
. PMID 1563350. - ↑ Thelen, M. P.; Venclovas, C.; Fidelis, K. (1999). "A sliding clamp model for the Rad1 family of cell cycle checkpoint proteins". Cell. 96 (6): 769–770. doi:10.1016/s0092-8674(00)80587-5. PMID 10102265.
- ↑ Walworth, N.; Davey, S.; Beach, D. (1993). "Fission yeast chkl protein kinase links the rad checkpoint pathway to cdc2". Nature. 363 (6427): 368–371. doi:10.1038/363368a0. PMID 8497322.
- ↑ Lee, J.; Kumagai, A.; Dunphy, W. G. (2001). "Positive regulation of Wee1 by Chk1 and 14-3-3 proteins". Molecular Biology of the Cell. 12 (3): 551–563. doi:10.1091/mbc.12.3.551. PMC 30963. PMID 11251070.
- ↑ Verkade, H. M.; Bugg, S. J.; Lindsay, H. D.; Carr, A. M.; O'Connell, M. J. (1999). "Rad18 is required for DNA repair and checkpoint responses in fission yeast". Molecular Biology of the Cell. 10 (9): 2905–2918. doi:10.1091/mbc.10.9.2905. PMC 25529. PMID 10473635.