Heme
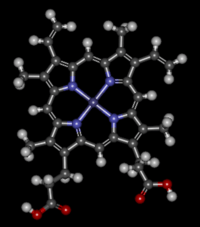
Heme or haem (from Greek αἷμα haima meaning blood) is a cofactor consisting of an Fe2+ (ferrous) ion contained in the centre of a large heterocyclic organic ring called a porphyrin, made up of four pyrrolic groups joined together by methine bridges. Not all porphyrins contain iron, but a substantial fraction of porphyrin-containing metalloproteins have heme as their prosthetic group; these are known as hemoproteins. Hemes are most commonly recognized as components of hemoglobin, the red pigment in blood, but are also found in a number of other biologically important hemoproteins such as myoglobin, cytochrome, catalase, heme peroxidase, and endothelial nitric oxide synthase.
Function
Hemoproteins have diverse biological functions including the transportation of diatomic gases, chemical catalysis, diatomic gas detection, and electron transfer. The heme ion serves as a source or sink of electrons during electron transfer or redox chemistry. In peroxidase reactions, the porphyrin molecule also serves as an electron source. In the transportation or detection of diatomic gases, the gas binds to the heme ion. During the detection of diatomic gases, the binding of the gas ligand to the heme ion induces conformational changes in the surrounding protein.
It has been speculated that the original evolutionary function of hemoproteins was electron transfer in primitive sulfur-based photosynthesis pathways in ancestral cyanobacteria-like organisms before the appearance of molecular oxygen.[1]
Hemoproteins achieve their remarkable functional diversity by modifying the environment of the heme macrocycle within the protein matrix. For example, the ability of hemoglobin to effectively deliver oxygen to tissues is due to specific amino acid residues located near the heme molecule. Hemoglobin binds oxygen in the lungs, where the pH is high and the CO2 concentration is low, and releases it in the tissues, where the situation is reversed. This phenomenon is known as the Bohr effect. The molecular mechanism behind this effect is the steric organization of the globin chain; a histidine residue, located adjacent to the heme group, becomes positively charged under acid (low pH) circumstances (which are caused by dissolved CO2 in working muscles, etc.), sterically releasing oxygen from the heme group.
Types
Major hemes
There are several biologically important kinds of heme:
| Heme A | Heme B | Heme C | Heme O | ||
|---|---|---|---|---|---|
| PubChem number | 7888115 | 444098 | 444125 | 6323367 | |
| Chemical formula | C49H56O6N4Fe | C34H32O4N4Fe | C34H36O4N4S2Fe | C49H58O5N4Fe | |
| Functional group at C3 |  |
–CH(OH)CH2Far | –CH=CH2 | –CH(cystein-S-yl)CH3 | –CH(OH)CH2Far |
| Functional group at C8 | –CH=CH2 | –CH=CH2 | –CH(cystein-S-yl)CH3 | –CH=CH2 | |
| Functional group at C18 | –CH=O | –CH3 | –CH3 | –CH3 | |
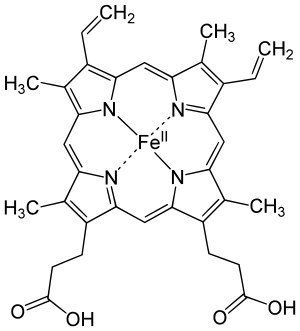
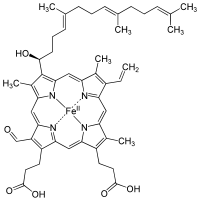
The most common type is heme B; other important types include heme A and heme C. Isolated hemes are commonly designated by capital letters while hemes bound to proteins are designated by lower case letters. Cytochrome a refers to the heme A in specific combination with membrane protein forming a portion of cytochrome c oxidase.
Other hemes
- The following carbon numbering system of porphyrins is an older numbering used by biochemists and not the 1–24 numbering system recommended by IUPAC which is shown in the table above.
- Heme l is the derivative of heme B which is covalently attached to the protein of lactoperoxidase, eosinophil peroxidase, and thyroid peroxidase. The addition of peroxide with the glutamyl-375 and aspartyl-225 of lactoperoxidase forms ester bonds between these amino acid residues and the heme 1- and 5-methyl groups, respectively.[4] Similar ester bonds with these two methyl groups are thought to form in eosinophil and thyroid peroxidases. Heme l is one important characteristic of animal peroxidases; plant peroxidases incorporate heme B. Lactoperoxidase and eosinophil peroxidase are protective enzymes responsible for the destruction of invading bacteria and virus. Thyroid peroxidase is the enzyme catalyzing the biosynthesis of the important thyroid hormones. Because lactoperoxidase destroys invading organisms in the lungs and excrement, it is thought to be an important protective enzyme.
- Heme m is the derivative of heme B covalently bound at the active site of myeloperoxidase. Heme m contains the two ester bonds at the heme 1- and 5-methyls as in heme l found in other mammalian peroxidases. In addition, a unique sulfonium ion linkage between the sulfur of a methionyl amino-acid residue and the heme 2-vinyl group is formed, giving this enzyme the unique capability of easily oxidizing chloride and bromide ions. Myeloperoxidase is present in mammalian neutrophils and is responsible for the destruction of invading bacteria and viruses. It also synthesizes hypobromite by "mistake" which is a known mutagenic compound.
- Heme D is another derivative of heme B, but in which the propionic acid side chain at the carbon of position 6, which is also hydroxylated, forms a γ-spirolactone. Ring III is also hydroxylated at position 5, in a conformation trans to the new lactone group.[5] Heme D is the site for oxygen reduction to water of many types of bacteria at low oxygen tension.
- Heme S is related to heme B by the having a formyl group at position 2 in place of the 2-vinyl group. Heme S is found in the hemoglobin of marine worms. The correct structures of heme B and heme S were first elucidated by German chemist Hans Fischer.
The names of cytochromes typically (but not always) reflect the kinds of hemes they contain: cytochrome a contains heme A, cytochrome c contains heme C, etc.
Synthesis
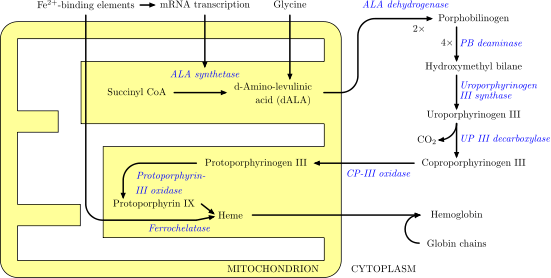
The enzymatic process that produces heme is properly called porphyrin synthesis, as all the intermediates are tetrapyrroles that are chemically classified as porphyrins. The process is highly conserved across biology. In humans, this pathway serves almost exclusively to form heme. In other species, it also produces similar substances such as cobalamin (vitamin B12).
The pathway is initiated by the synthesis of D-aminolevulinic acid (dALA or δALA) from the amino acid glycine and succinyl-CoA from the citric acid cycle (Krebs cycle). The rate-limiting enzyme responsible for this reaction, ALA synthase, is negatively regulated by glucose and heme concentration. Mechanism of inhibition of ALAs by heme or hemin is by decreasing stability of mRNA synthesis and by decreasing the intake of mRNA in the mitochondria. This mechanism is of therapeutic importance: infusion of heme arginate or hematin and glucose can abort attacks of acute intermittent porphyria in patients with an inborn error of metabolism of this process, by reducing transcription of ALA synthase.[6]
The organs mainly involved in heme synthesis are the liver (in which the rate of synthesis is highly variable, depending on the systemic heme pool) and the bone marrow (in which rate of synthesis of Heme is relatively constant and depends on the production of globin chain), although every cell requires heme to function properly. However, due to its toxic properties, proteins such as Hemopexin (Hx) are required to help maintain physiological stores of iron in order for them to be used in synthesis. [7] Heme is seen as an intermediate molecule in catabolism of hemoglobin in the process of bilirubin metabolism. Defects in various enzymes in synthesis of heme can lead to group of disorder called porphyrias, these include acute intermittent porphyria, congenital erythropoetic porphyria, porphyria cutanea tarda, hereditary coproporphyria, variegate porphyria, erythropoietic protoporphyria.
Degradation
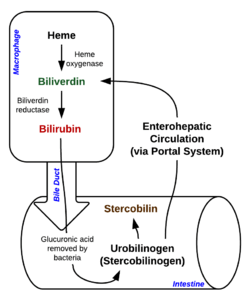
Degradation begins inside macrophages of the spleen, which remove old and damaged (senescent) erythrocytes from the circulation. In the first step, heme is converted to biliverdin by the enzyme heme oxygenase (HMOX). NADPH is used as the reducing agent, molecular oxygen enters the reaction, carbon monoxide (CO) is produced and the iron is released from the molecule as the ferrous ion (Fe2+).[8] CO acts as a cellular messenger and functions in vasodilation.
In addition, heme degradation appears to be an evolutionarily-conserved response to oxidative stress. Briefly, when cells are exposed to free radicals, there is a rapid induction of the expression of the stress-responsive heme oxygenase-1 (HMOX1) isoenzyme that catabolizes heme (see below). The reason why cells must increase exponentially their capability to degrade heme in response to oxidative stress remains unclear but this appears to be part of a cytoprotective response that avoids the deleterious effects of free heme. When large amounts of free heme accumulates, the heme detoxification/degradation systems get overwhelmed, enabling heme to exert its damaging effects.[9]
| heme | heme oxygenase-1 | biliverdin + Fe2+ | |
| |
 | ||
| H+ + NADPH + O2 | NADP+ + CO | ||
| | |||
In the second reaction, biliverdin is converted to bilirubin by biliverdin reductase (BVR):
| biliverdin | biliverdin reductase | bilirubin | |
| |
 | ||
| H+ + NADPH | NADP+ | ||
| | |||
Bilirubin is transported into the liver by facilitated diffusion bound to a protein (serum albumin), where it is conjugated with glucuronic acid to become more water-soluble. The reaction is catalyzed by the enzyme UDP-glucuronosyltransferase.
| bilirubin | UDP-glucuronosyltransferase | bilirubin diglucuronide | |
| |
 | ||
| 2 UDP-glucuronide | 2 UMP + 2 Pi | ||
| | |||
This form of bilirubin is excreted from the liver in bile. Excretion of bilirubin from liver to biliary canaliculi is an active, energy dependent and rate limiting process. The intestinal bacteria deconjugate bilirubin diglucuronide and convert bilirubin to urobilinogens. Some urobilinogen is absorbed by intestinal cells and transported into the kidneys and excreted with urine (urobilin, which is the product of oxidation of urobilinogen, is responsible for the yellow colour of urine). The remainder travels down the digestive tract and is converted to stercobilinogen. This is oxidized to stercobilin, which is excreted and is responsible for the color of feces.
In health and disease
Under homeostasis, the reactivity of heme is controlled by its insertion into the “heme pockets” of hemoproteins. Under oxidative stress however, some hemoproteins, e.g. hemoglobin, can release their heme prosthetic groups.[10][11] The non-protein-bound (free) heme produced in this manner becomes highly cytotoxic, most probably due to the iron atom contained within its protoporphyrin IX ring, which can act as a Fenton's reagent to catalyze in an unfettered manner the production of free radicals.[12] It catalyzes the oxidation and aggregation of protein, the formation of cytotoxic lipid peroxide via lipid peroxidation and damages DNA through oxidative stress. Due to its lipophilic properties, it impairs lipid bilayers in organelles such as mitochondria and nuclei. [13]These properties of free heme can sensitize a variety of cell types to undergo programmed cell death in response to pro-inflammatory agonists, a deleterious effect that plays an important role in the pathogenesis of certain inflammatory diseases such as malaria[14] and sepsis.[15] There is an association between high intake of heme iron sourced from meat and increased risk of colon cancer;[16] however, there is no solid evidence that this is a causal relationship. The heme content of red meat is 10-fold higher than that of white meat such as chicken.[17]
Genes
The following genes are part of the chemical pathway for making heme:
- ALAD: aminolevulinic acid, δ-, dehydratase (deficiency causes ala-dehydratase deficiency porphyria)[18]
- ALAS1: aminolevulinate, δ-, synthase 1
- ALAS2: aminolevulinate, δ-, synthase 2 (deficiency causes sideroblastic/hypochromic anemia)
- CPOX: coproporphyrinogen oxidase (deficiency causes hereditary coproporphyria)[19]
- FECH: ferrochelatase (protoporphyria)
- HMBS: hydroxymethylbilane synthase (deficiency causes acute intermittent porphyria)[20]
- PPOX: protoporphyrinogen oxidase (deficiency causes variegate porphyria)[21]
- UROD: uroporphyrinogen decarboxylase (deficiency causes porphyria cutanea tarda)[22]
- UROS: uroporphyrinogen III synthase (deficiency causes congenital erythropoietic porphyria)
See also
Notes and references
- ↑ Hardison, R. (1999). "The Evolution of Hemoglobin: Studies of a very ancient protein suggest that changes in gene regulation are an important part of the evolutionary story". American Scientist. 87 (2): 126.
- ↑ Caughey, Winslow S.; et al. (1975). "Heme A of Cytochrome c Oxidase: Structure and properties: comparisons with hemes B, C, and S and derivatives". J. Biol. Chem. 250 (19): 7602–7622. PMID 170266.
- ↑ Hegg, Eric L.; et al. (2004). "Heme A Synthase Does Not Incorporate Molecular Oxygen into the Formyl Group of Heme A". Biochemistry. 43 (27): 8616–8624. doi:10.1021/bi049056m. PMID 15236569.
- ↑ Rae, T.; Goff, H. (1998). "The heme prosthetic group of lactoperoxidase. Structural characteristics of heme l and heme l-peptides". The Journal of Biological Chemistry. 273 (43): 27968–27977. doi:10.1074/jbc.273.43.27968. PMID 9774411.
- ↑ Murshudov, G.; Grebenko, A.; Barynin, V.; Dauter, Z.; Wilson, K.; Vainshtein, B.; Melik-Adamyan, W.; Bravo, J.; Ferrán, J.; Ferrer, J. C.; Switala, J.; Loewen, P. C.; Fita, I. (1996). "Structure of the heme d of Penicillium vitale and Escherichia coli catalases". The Journal of Biological Chemistry. 271 (15): 8863–8868. doi:10.1074/jbc.271.15.8863. PMID 8621527.
- ↑ http://escholarship.umassmed.edu/gsbs_diss/121/
- ↑ Kumar, Sanjay; Bandyopadhyay, Uday (July 2005). "Free heme toxicity and its detoxification systems in human". Toxicology Letters. 157 (3): 175–188.
- ↑ Lehninger's Principles of Biochemistry (5th ed.). New York: W. H. Freeman and Company. 2008. p. 876. ISBN 978-0-7167-7108-1.
- ↑ Kumar, Sanjay; Bandyopadhyay, Uday (July 2005). "Free heme toxicity and its detoxification systems in human". Toxicology Letters. 157 (3): 175–188.
- ↑ Bunn, H. F.; Jandl, J. H. (Sep 1966). "Exchange of heme among hemoglobin molecules". Proc. Natl. Acad. Sci. USA. 56 (3): 974–978. doi:10.1073/pnas.56.3.974. PMID 5230192.
- ↑ Smith, M. L.; Paul, J.; Ohlsson, P. I.; Hjortsberg, K.; Paul, K. G. (Feb 1991). "Heme-protein fission under nondenaturing conditions". Proc. Natl. Acad. Sci. USA. 88 (3): 882–886. Bibcode:1991PNAS...88..882S. doi:10.1073/pnas.88.3.882. PMID 1846966.
- ↑ Everse, J.; Hsia, N. (1197). "The toxicities of native and modified hemoglobins". Free Radical Biology and Medicine. 22 (6): 1075–1099. doi:10.1016/S0891-5849(96)00499-6. PMID 9034247.
- ↑ Kumar, Sanjay; Bandyopadhyay, Uday (July 2005). "Free heme toxicity and its detoxification systems in humans". Toxicology Letters. 157 (3): 175–188.
- ↑ Pamplona, A.; Ferreira, A.; Balla, J.; Jeney, V.; Balla, G.; Epiphanio, S.; Chora, A.; Rodrigues, C. D.; Gregoire, I. P.; Cunha-Rodrigues, M.; Portugal, S.; Soares, M. P.; Mota, M. M. (Jun 2007). "Heme oxygenase-1 and carbon monoxide suppress the pathogenesis of experimental cerebral malaria". Nature Medicine. 13 (6): 703–710. doi:10.1038/nm1586. PMID 17496899.
- ↑ Larsen, R.; Gozzelino, R.; Jeney, V.; Tokaji, L.; Bozza, F. A.; Japiassú, A. M.; Bonaparte, D.; Cavalcante, M. M.; Chora, A.; Ferreira, A.; Marguti, I.; Cardoso, S.; Sepúlveda, N.; Smith, A.; Soares, M. P. (2010). "A central role for free heme in the pathogenesis of severe sepsis". Science Translational Medicine. 2 (51): 51ra71. doi:10.1126/scitranslmed.3001118. PMID 20881280.
- ↑ Bastide, N. M.; Pierre, F. H.; Corpet, D. E. (2011). "Heme iron from meat and risk of colorectal cancer: a meta-analysis and a review of the mechanisms involved" (PDF). Cancer Prev. Res. 4 (2): 177–184. doi:10.1158/1940-6207.CAPR-10-0113. PMID 21209396.
- ↑ http://cancerpreventionresearch.aacrjournals.org/content/4/2/177.full
- ↑ Plewinska, Magdalena; Thunell, Stig; Holmberg, Lars; Wetmur, James; Desnick, Robert (1991). "delta-Aminolevulinate dehydratase deficient porphyria: identification of the molecular lesions in a severely affected homozygote". American Journal of Human Genetics. 49 (1): 167–174. PMC 1683193
 . PMID 2063868.
. PMID 2063868. - ↑ Aurizi, C.; Lupia Palmieri, G.; Barbieri, L.; Macri, A.; Sorge, F.; Usai, G.; Biolcati, G. (February 2009). "Four novel mutations of the coproporphyrinogen III oxidase gene". Cellular and Molecular Biology. 55 (1): 8–15.
- ↑ Bustad, H. J.; Vorland, M.; Ronneseth, E.; Sandberg, S.; Martinez, A.; Toska, K. (August 8, 2013). "Conformational stability and activity analysis of two hydroxymethylbilane synthase mutants, K132N and V215E, with different phenotypic association with acute intermittent porphyria". Bioscience Reports. 33 (4): 617–626. doi:10.1042/BSR20130045.
- ↑ Martinez di Montemuros, F.; Di Pierro, E.; Patti, E.; Tavazzi, D.; Danielli, M. G.; Biolcati, G.; Rocchi, E.; Cappllini, M. D. (December 2002). "Molecular characterization of porphyrias in Italy: a diagnostic flow-chart". 48 (8): 867–876.
- ↑ Badenas, C.; To Figueras, J.; Phillips, J. D.; Warby, C. A.; Muñoz, C.; Herrero, C. (April 2009). "Identification and characterization of novel uroporphyrinogen decarboxylase gene mutations in a large series of porphyria cutanea tarda patients and relatives". Clinical Genetics. 75 (4): 346–353. doi:10.1111/j.1399-0004.2009.01153.x. PMID 19419417.