Mechanistic organic photochemistry
Mechanistic organic photochemistry is that aspect of organic photochemistry which seeks to explain the mechanisms of organic photochemical reactions.[1][2] The absorption of ultraviolet light by organic molecules very often leads to reactions. In the earliest days sunlight was employed while in more modern times ultraviolet lamps are employed. Organic photochemistry has proven to be a very useful synthetic tool. Complex organic products can be obtained simply. Over the last century and earlier an immense number of photochemical reactions have been uncovered. In modern times the field is quite well understood and is used in organic synthesis and industrially. The utility of organic photochemistry has arisen only by virtue of the available mechanistic treatment; reactions which appear unlikely in ground-state understanding become understandable and accessible in terms of electronic excited-state consideration.
History
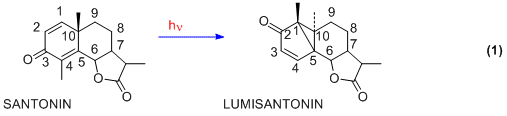
One of the earliest photochemical studies dealt with the natural product santonin. In the 19th century it had been observed by Ciamician that in Italian sunlight santonin gave several photoproducts.[3][4] The structure of santonin was first correctly described by Clemo and Hayworth in 1929.[5] The initial photoproduct obtained from santonin is lumisantonin.[6] As depicted in Eqn. 1, the photoreaction involves a rearrangement. Using steroid numbering, we note that the C-3 carbonyl group has moved to C-2, the C-4 methyl has moved to C-1, and the C-10 carbon has been inverted.
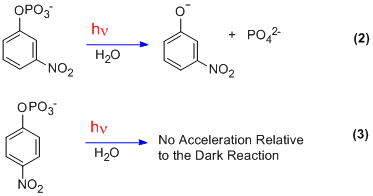
A comparatively bizarre example was uncovered by Egbert Havinga in 1956.[7] The curious result was activation on photolysis by a meta nitro group in contrast to the usual activation by ortho and para groups.
Over the decades, many interesting but puzzling organic photochemical reactions were discovered that did not proceed by ordinary organic ground state processes. Rather, they arose from the excited states of electrons in the compounds. The real problem was that, at the time, organic chemists were not versed in quantum mechanics and physical chemists were not versed in organic chemistry. Real mechanistic treatments were not possible.
Starting in 1961 it was found that one could understand organic photochemical reactions in the context of the relevant excited states.[8][9][10] One example is the n-pi* excitation of mono-carbonyl compounds, the simplest being that of formaldehyde. The structure was first described by Mulliken.[11] The three-dimensional representation (top drawing) is simplified in the second line using a two-dimensional representation, which facilitates arrow pushing.[9][10]

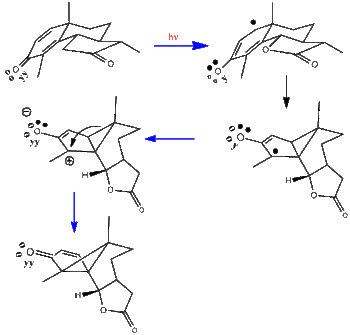
In this early research simple Hückel computations were used to get excited state electron densities and bond-orders.[9][10] The stereochemistry in Scheme 1 is shown three-dimensionally. The Hückel computations revealed that the beta-carbons (i.e. C2 and C5) of the cyclohexadienone ring had a large bond-order. As seen in the scheme a beta-beta bond is formed. Subsequent to this, radiationless decay leads to a zwitterion ground state. The final rearrangement leads to lumisantonin as can be discerned by comparing the three-dimensional drawing with the earlier two-dimensional representation.
Scheme 1. The santonin to lumisantonin rearrangement in three-dimensions
4,4-Diphenylcyclohexadienone rearrangement
Quite parallel to the santonin to lumisantonin example is the rearrangement of 4,4-diphenylcyclohexadienone[10] Here the n-pi* triplet excited state undergoes the same beta-beta bonding. This is followed by intersystem crossing (i.e. ISC) to form the singlet ground state which is seen to be a zwitterion. The final step is the rearrangement to the bicyclic photoproduct. The reaction is termed the type A cyclohexadienone rearrangement.

The contrasting case of 4,4-diphenylcyclohexenone, one double bond missing
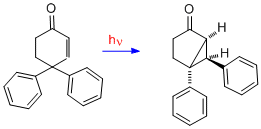
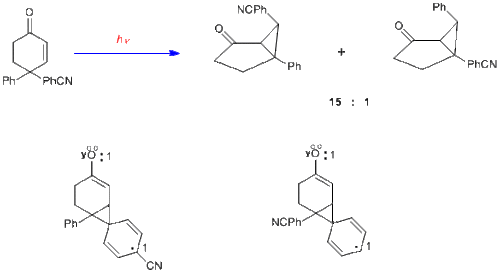
To provide further evidence on the mechanism of the dienone in which there is bonding between the two double bonds, the case of 4,4-diphenylcyclohexenone is presented here. It is seen that the rearrangement is quite different; thus two double bonds are required for a type A rearrangement. With one double bond one of the phenyl groups, originally at C-4, has migrated to C-3 (i.e. the beta carbon).[12]
It is of considerable interest that when one of the aryl groups has a para-cyano or para-methoxy group, that substituted aryl group migrates in preference.[13] Inspection of the alternative phenonium-type species, in which an aryl group has begun to migrate to the beta-carbon, reveals the greater electron delocalization with a substituent para on the migrating aryl group and thus a more stabilized pathway.
Pi-pi* reactivity
Still another type of photochemical reaction is the di-pi-methane rearrangement.[14] Two further early examples were the rearrangement of 1,1,5,5-tetraphenyl-3,3-dimethyl-1,4-pentadiene (the "Mariano" molecule)[15] and the rearrangement of barrelene to semibullvalene.[16] We note that, in contrast to the cyclohexadienone reactions which used n-pi* excited states, the di-pi-methane rearrangements utilize pi-pi* excited states.
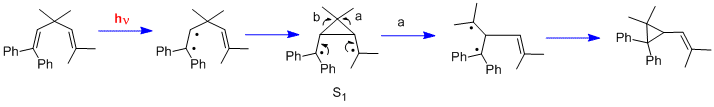
Parallel studies on multiplicity; the role of triplets
Parallel to the structural studies of the Zimmerman group as described above, the Caltech group with George Hammond pursued the role of multiplicity on reactivity. The importance of triplet excited species was emphasized. The triplets tend to be longer lived than singlets and of lower energy than the singlet of the same configuration. Hammond had summarized the energies of triplets of the more common molecules. He noted that triplets may arise from (A) conversion of the initially formed singlets or by (B) interaction with a higher energy triplet (sensitization). Another contribution of Hammond was the determination of triplet reaction rates. Finally, with low-energy triplets present it was shown possible to quench a triplet reaction.[17] The groundwork had been laid by very earlier studies in Russia by Terenin and Ermolaev, who demonstrated intermolecular triplet transfer at low temperatures and described the kinetics.[18]
Further on the subsequent years
After the basics had been established, organic photochemistry seemed to accelerate exponentially. At Caltech, George Hammond had a remarkable group of researchers including Nicholas Turro, Angelo Lamola, Peter Leermakers, Jack Saltiel, Robert Liu and a number of others.[19] In particular, Nicholas Turro is the most prolific and highly cited of the pioneers of modern organic photochemistry. His textbook, Modern Molecular Photochemistry, is considered the "Bible" of the field. A highly influential aspect of Turro's work involved applying the paradigms of photochemistry to more complex environments relative to small molecule solution-phase experiments including supramolecular systems, interfaces, restricted spaces and biological systems.
Common organic photochemical reactions
Among the more common organic photochemical reactions there are the Norrish Type I, the Norrish Type II, the racemization of optically active biphenyls, the type A cyclohexadienone rearrangement, the type B cyclohexenone rearrangement, the di-pi-methane rearrangement, the type B bicyclo[3.1.0]hexanone rearrangement to phenols, photochemical electrocyclic processes, the rearrangement of epoxyketones to beta-diketones, ring opening of cyclopropyl ketones, heterolysis of 3,5-dimethoxylbenzylic derivatives, and photochemical cyclizations of dienes. Some of these have been described above and with this being an encyclopedic survey only a selected few are considered here.
References
- ↑ P. Klán, J. Wirz Photochemistry of Organic Compounds: From Concepts to Practice. Wiley, Chichester, 2009, ISBN 978-1405190886.
- ↑ N. J. Turro, V. Ramamurthy, J. C. Scaiano Modern Molecular Photochemistry of Organic Molecules. University Science Books, Sausalito, 2010, ISBN 978-1891389252.
- ↑ Roth, H. D., Angew. Chem. Int. Ed. 1989, 1l93-1207
- ↑ Woodward, R. B.; Brutschy, F. J.; Baer, H., J. Am. Chem. Soc.. 1948, 70 4216-4221.
- ↑ Clemo, G. R.;Haworth, R. D., J. Chem Soc., 1929, 2369-2387. Clemo, G. R.; Haworth, R. D.; Walton, E. J. Chcm. ,Soc., 1930, 1110-1115. Clemo, G. R.; Haworth, R. D.; Walton, E. J. Chcm. ,Soc., 1930, 2368-2387
- ↑ Barton, D. H. R; De Mayo, P.; Shafiq, M., J. Chem. Soc., 1958, 140-145. Barton. D. H. R.; P. T. Gillam, P. T., 1960, J. Chem. Soc., 4596-4599.
- ↑ Havinga, E.; de Jongh, R. O.; Dorst, W.; Rec. Trav. Chim., 1956, 76, 378-383.
- ↑ "The Photochemical Rearrangement of 4,4-Diphenylcyclohexadienone. Paper I on a General Theory of Photochemical Reactions," Zimmerman, H. E.; Schuster, D. I. J. Am. Chem. Soc., 1961, 83, 4486-4487.
- 1 2 3 "A Mechanistic Approach to Organic Photochemistry," Zimmerman, H. E., Seventeenth National Organic Symposium of the Am. Chem. Soc., Abstracts, Bloomington, Indiana, 1961, pgs. 31-41. doi:10.1021/cen-v039n016.p084
- 1 2 3 4 Zimmerman, Howard E.; David I. Schuster (1962). "A New Approach to Mechanistic Organic Photochemistry. IV. Photochemical Rearrangements of 4,4-Diphenylcyclohexadienone". J.A.C.S. A.C.S. 84 (23): 4527–4540. doi:10.1021/ja00882a032.
- ↑ "Electronic Structures of Molecues. Aldehydes, Ketones and Related Molecules", Mulliken, R. S., J. Chem. Phys. 1935, 3, 564-573.
- ↑ "Mechanistic and Exploratory Organic Photochemistry, IX. Phenyl Migration in the Irradiation of 4.4-Diphenylcyclohexenone," Zimmerman, H. E.; Wilson, J. W. J. Am. Chem. Soc., 1964, 86, 4036-4042.
- ↑ "Photochemical Migratory Aptitudes in Cyclohexenones. Mechanistic and Exploratory Organic Photochemistry. XXIII," Zimmerman, H. E.; Rieke, R. D.; Scheffer, J. R. J. Am. Chem. Soc., 1967, 89, 2033-2047.
- ↑ "Unsymmetrical Substitution and the Direction of the Di-pi-Methane Rearrangement; Mechanistic and Exploratory Organic Photochemistry. LVI," Zimmerman, H. E.; Pratt, A. C. J. Am. Chem. Soc., 1970, 92, 6259-6267
- ↑ "The Di-pi-Methane Rearrangement. Interaction of Electronically Excited Vinyl Chromophores. Zimmerman, H. E.; Mariano, P. S. J. Am. Chem. Soc., 1969, 91, 1718-1727.
- ↑ Zimmerman, H. E.; Grunewald, G. L. (1966). "The Chemistry of Barrelene. III. A Unique Photoisomerization to Semibullvalene". J. Am. Chem. Soc. 88 (1): 183–184. doi:10.1021/ja009
- ↑ Hammond, G. S., Seventeenth National Organic Symposium of the Am. Chem. Soc., Abstracts, Bloomington, Indiana, 1961, pgs 48–58
- ↑ "Terenin, A.; Ermolaev, V. Sensitized Phosphorescence in Organic Solutions at Low Temperature; Energy Transfer Between Triplet States", Trans. Faraday Soc., 1956, 52, 1042–1052.
- ↑ For example Mechanisms of photochemical reactions in solution. XXII. Photochemical cis-trans isomerization. Hammond, G. S.; Saltiel, J.; Lamola, A. A.; Turro, N. J.; Bradshaw, J. S.; Cowan, D. O.; Counsell, R. C.; Vogt, V.; Dalton, C., J. Amer. Chem. Soc. 1964, 86(16), 3197-3217.