Microglia
| Alternate Names | Microgliocyte, Gitter cell, Hortega Cell |
|---|---|
| Embryological Origin | Yolk-sac derived erythromyeloid progenitors |
| Antecedent Cell(s) | Hematopoietic stem cells, monocyte |
| System | Central nervous system |
| Location(s) | Brain and spinal cord |
| Longevitiy | Up to lifetime of organism[1] |
| Function(s) | Immune regulation, synaptic and neuronal pruning, extracellular signaling |
| Cytokine(s) | IFN-γ, IL-1α, IL-1β, IL-6,[2] IL-8, IL-18,[3] TNF-α[2] |
| Stains | CD68, Iba1, lectin proteins GS-1, RCA, WGA, and ConA,[4] LN3, TSPO[5] |
Microglia are a type of glial cell located throughout the brain and spinal cord.[6] Microglia account for 10–15% of all cells found within the brain.[7] As the resident macrophage cells, they act as the first and main form of active immune defense in the central nervous system (CNS).[8] Microglia (and other glia including astrocytes) are distributed in large non-overlapping regions throughout the CNS.[9][10] Microglia are key cells in overall brain maintenance — they are constantly scavenging the CNS for plaques, damaged or unnecessary neurons and synapses, and infectious agents.[11] Since these processes must be efficient to prevent potentially fatal damage, microglia are extremely sensitive to even small pathological changes in the CNS.[12] This sensitivity is achieved in part by the presence of unique potassium channels that respond to even small changes in extracellular potassium.[11]
The brain and spinal cord, which make up the CNS, are not usually accessed directly by pathogenic factors in the body's circulation due to a series of endothelial cells known as the blood–brain barrier, or BBB. The BBB prevents most infections from reaching the vulnerable nervous tissue. In the case where infectious agents are directly introduced to the brain or cross the blood–brain barrier, microglial cells must react quickly to decrease inflammation and destroy the infectious agents before they damage the sensitive neural tissue. Due to the unavailability of antibodies from the rest of the body (few antibodies are small enough to cross the blood–brain barrier), microglia must be able to recognize foreign bodies, swallow them, and act as antigen-presenting cells activating T-cells.
-

Microglia – ramified form from rat cortex before traumatic brain injury (lectin staining with HRP)
-

Microglia/macrophage – activated form from rat cortex after traumatic brain injury (lectin staining with HRP)
-

Rat microglia grown in tissue culture in green, along with nerve fiber processes shown in red
Origin
Microglial cells differentiate in the bone marrow from hematopoietic stem cells, the progenitors of all blood cells. During hematopoiesis, some of these stem cells differentiate into monocytes and travel from the bone marrow to the brain, where they settle and further differentiate into microglia. However, recent studies indicate microglia originate in the yolk sac during a remarkably restricted period and populate the brain mesenchyme.[13]
Monocytes can also differentiate into myeloid dendritic cells and macrophages in the peripheral systems. Like macrophages in the rest of the body, microglia use phagocytic and cytotoxic mechanisms to destroy foreign materials. Microglia and macrophages both contribute to the immune response by acting as antigen presenting cells, as well as promoting inflammation and homeostatic mechanisms within the body by secreting cytokines and other signaling molecules.
In their downregulated form, microglia lack the MHC class I/MHC class II proteins, IFN-γ cytokines, CD45 antigens, and many other surface receptors required to act in the antigen-presenting, phagocytic, and cytotoxic roles that hallmark normal macrophages. Microglia also differ from macrophages in that they are much more tightly regulated spatially and temporally in order to maintain a precise immune response.[14]
Another difference between microglia and other cells that differentiate from myeloid progenitor cells is the turnover rate. Macrophages and dendritic cells are constantly being used up and replaced by myeloid progenitor cells which differentiate into the needed type. Due to the blood–brain barrier, it would be fairly difficult for the body to constantly replace microglia. Therefore, instead of constantly being replaced with myeloid progenitor cells, the microglia maintain their status quo while in their quiescent state, and then, when they are activated, they rapidly proliferate in order to keep their numbers up. Bone chimera studies have shown, however, that in cases of extreme infection the blood–brain barrier will weaken, and microglia will be replaced with haematogenous, marrow-derived cells, namely myeloid progenitor cells and macrophages. Once the infection has decreased the disconnect between peripheral and central systems is reestablished and only microglia are present for the recovery and regrowth period.[15]
History
The ability to view and characterize different neural cells including microglia began in 1880 when Nissl staining was developed by Franz Nissl. Franz Nissl and F. Robertson first described microglial cells during their histology experiments. The cell staining techniques in the 1880s showed that microglia are related to macrophages. The activation of microglia and formation of ramified microglial clusters was first noted by Victor Babeş while studying a rabies case in 1897. Babeş noted the cells were found in a variety of viral brain infections but did not know what the clusters of microglia he saw were.[16] Pío del Río Hortega, a student of Santiago Ramón y Cajal, first called the cells "microglia" around 1920. He went on to characterize microglial response to brain lesions in 1927 and note the "fountains of microglia" present in the corpus callosum and other perinatal white matter areas in 1932. After many years of research Rio-Hortega became generally considered as the "Father of Microglia."[17][18] For a long period of time little improvement was made in our knowledge of microglia. Then, in 1988, Hickey and Kimura showed that perivascular microglial cells are bone-marrow derived, and express high levels of MHC class II proteins used for antigen presentation. This confirmed Pio Del Rio-Hortega's postulate that microglial cells functioned similarly to macrophages by performing phagocytosis and antigen presentation.
Forms
Microglial cells are extremely plastic, and undergo a variety of structural changes based on location and system needs. This level of plasticity is required to fulfill the vast variety of functions that microglia perform. The ability to transform distinguishes microglia from macrophages, which must be replaced on a regular basis, and provides them the ability to defend the CNS on extremely short notice without causing immunological disturbance.[11] Microglia adopt a specific form, or phenotype, in response to the local conditions and chemical signals they have detected.[19]
Ramified
This form of microglial cell is commonly found at specific locations throughout the entire brain and spinal cord in the absence of foreign material or dying cells. This "resting" form of microglia is composed of long branching processes and a small cellular body. Unlike the amoeboid forms of microglia, the cell body of the ramified form remains in place while its branches are constantly moving and surveying the surrounding area. The branches are very sensitive to small changes in physiological condition and require very specific culture conditions to observe in vitro.[19]
Unlike activated or ameboid microglia, ramified microglia do not phagocytose cells and secrete fewer immunomolecules (including the MHC class I/II proteins). Microglia in this state are able to search for and identify immune threats while maintaining homeostasis in the CNS.[14][20][21] Although this is considered the resting state, microglia in this form are still extremely active in chemically surveying the environment. Ramified microglia can be transformed into activated form at any time in response to injury or threat.[19]
Reactive (Activated)
Although historically frequently used, the term "activated" microglia should be replaced by "reactive" microglia.[22] Indeed apparently quiescent microglia are not devoid of active functions and the "activation" term is misleading as it tends to indicate an "all or nothing" polarization of cell reactivity. The marker Iba1, which is upregulated in reactive microglia, is often used to visualize these cells.
Non-phagocytic
This state is actually part of a graded response as microglia move from their ramified form to their fully active phagocytic form. Microglia can be activated by a variety of factors including: glutamate receptor agonists, pro-inflammatory cytokines, cell necrosis factors, lipopolysaccharide, and changes in extracellular potassium (indicative of ruptured cells). Once activated the cells undergo several key morphological changes including the thickening and retraction of branches, uptake of MHC class I/II proteins, expression of immunomolecules, secretion of cytotoxic factors, secretion of recruitment molecules, and secretion of pro-inflammatory signaling molecules (resulting in a pro-inflammation signal cascade). Activated non-phagocytic microglia generally appear as "bushy," "rods," or small ameboids depending on how far along the ramified to full phagocytic transformation continuum they are. In addition, the microglia also undergo rapid proliferation in order to increase their numbers. From a strictly morphological perspective, the variation in microglial form along the continuum is associated with changing morphological complexity and can be quantitated using the methods of fractal analysis, which have proven sensitive to even subtle, visually undetectable changes associated with different morphologies in different pathological states.[11][14][20][23]
Phagocytic
Activated phagocytic microglia are the maximally immune responsive form of microglia. These cells generally take on a large, ameboid shape, although some variance has been observed. In addition to having the antigen presenting, cytotoxic and inflammatory mediating signaling of activated non-phagocytic microglia, they are also able to phagocytose foreign materials and display the resulting immunomolecules for T-cell activation. Phagocytic microglia travel to the site of the injury, engulf the offending material, and secrete pro-inflammatory factors to promote more cells to proliferate and do the same. Activated phagocytic microglia also interact with astrocytes and neural cells to fight off the infection as quickly as possible with minimal damage to the healthy brain cells.[11][14]
Amoeboid
This shape allows the microglial free movement throughout the neural tissue, which allows it to fulfill its role as a scavenger cell. Amoeboid microglia are able to phagocytose debris, but do not fulfill the same antigen-presenting and inflammatory roles as activated microglia. Amoeboid microglia are especially prevalent during the development and rewiring of the brain, when there are large amounts of extracellular debris and apoptotic cells to remove. This form of microglial cell is found mainly within the perinatal white matter areas in the corpus callosum known as the "Fountains of Microglia."[11][20][24]
Gitter cells
Gitter cells are the eventual result of microglial cell's phagocytosis of infectious material or cellular debris. Eventually, after engulfing a certain amount of material, the phagocytic microglia becomes unable to phagocytose any further materials. The resulting cellular mass is known as a granular corpuscle, named for its ‘grainy' appearance. By looking at tissues stained to reveal gitter cells, pathologists can see post-infection areas that have healed.[25]
Perivascular
Unlike the other types of microglia mentioned above, "perivascular" microglia refers to the location of the cell rather than its form/function. Perivascular microglia are mainly found encased within the walls of the basal lamina. They perform normal microglial functions, but unlike normal microglia they are replaced by bone marrow derived precursor cells on a regular basis and express MHC class II antigens regardless of the outside environment. Perivascular microglia also react strongly to macrophage differentiation antigens.[11] These microglia have been shown to be essential to repair of vascular walls, as shown by Ritter's experiments and observations on ischemic retinopathy. Perivascular microglia promote endothelial cell proliferation allowing new vessels to be formed and damaged vessels to be repaired. During repair and development, myeloid recruitment and differentiation into microglial cells is highly accelerated to accomplish these tasks.[13]
Juxtavascular
Like perivascular microglia, juxtavascular microglia can be distinguished mainly by their location. Juxtavascular microglia are found making direct contact with the basal lamina wall of blood vessels but are not found within the walls. Like perivascular cells, they express MHC class II proteins even at low levels of inflammatory cytokine activity. Unlike perivascular cells, but similar to resident microglia, juxtavascular microglia do not exhibit rapid turnover or replacement with myeloid precursor cells on a regular basis.[11]
Functions
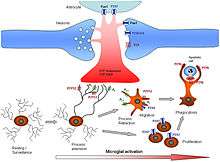
Microglial cells fulfill a variety of different tasks within the CNS mainly related to both immune response and maintaining homeostasis. The following are some of the major known functions carried out by these cells.
Scavenging
In addition to being very sensitive to small changes in their environment, each microglial cell also physically surveys its domain on a regular basis. This action is carried out in the ameboid and resting states. While moving through its set region, if the microglial cell finds any foreign material, damaged cells, apoptotic cells, neurofibrillary tangles, DNA fragments, or plaques it will activate and phagocytose the material or cell. In this manner microglial cells also act as "housekeepers", cleaning up random cellular debris.[14] During developmental wiring of the brain, microglial cells play a large role regulating numbers of neural precursor cells and removing apoptotic neurons. They also engulf synapses to regulate synaptic numbers. Post development, the majority of dead or apoptotic cells are found in the cerebral cortex and the subcortical white matter. This may explain why the majority of ameboid microglial cells are found within the "fountains of microglia" in the cerebral cortex.[24]
Phagocytosis
The main role of microglia, phagocytosis, involves the engulfing of various materials. Engulfed materials generally consist of cellular debris, lipids, and apoptotic cells in the non-inflamed state, and invading virus, bacteria, or other foreign materials in the inflamed state. Once the microglial cell is "full" it stops phagocytic activity and changes into a relatively non-reactive gitter cell.
Extracellular signaling
A large part of microglial cell's role in the brain is maintaining homeostasis in non-infected regions and promoting inflammation in infected or damaged tissue. Microglia accomplish this through an extremely complicated series of extracellular signaling molecules which allow them to communicate with other microglia, astrocytes, nerves, T-cells, and myeloid progenitor cells. As mentioned above the cytokine IFN-γ can be used to activate microglial cells. In addition, after becoming activated with IFN-γ, microglia also release more IFN-γ into the extracellular space. This activates more microglia and starts a cytokine induced activation cascade rapidly activating all nearby microglia. Microglia-produced TNF-α causes neural tissue to undergo apoptosis and increases inflammation. IL-8 promotes B-cell growth and differentiation, allowing it to assist microglia in fighting infection. Another cytokine, IL-1, inhibits the cytokines IL-10 and TGF-β, which downregulate antigen presentation and pro-inflammatory signaling. Additional dendritic cells and T-cells are recruited to the site of injury through the microglial production of the chemotactic molecules like MDC, IL-8, and MIP-3β. Finally, PGE2 and other prostanoids prevent chronic inflammation by inhibiting microglial pro-inflammatory response and downregulating Th1 (T-helper cell) response.[14]
Antigen presentation
As mentioned above, resident non-activated microglia act as poor antigen presenting cells due to their lack of MHC class I/II proteins. Upon activation they rapidly uptake MHC class I/II proteins and quickly become efficient antigen presenters. In some cases, microglia can also be activated by IFN-γ to present antigens, but do not function as effectively as if they had undergone uptake of MHC class I/II proteins. During inflammation, T-cells cross the blood–brain barrier thanks to specialized surface markers and then directly bind to microglia in order to receive antigens. Once they have been presented with antigens, T-cells go on to fulfill a variety of roles including pro-inflammatory recruitment, formation of immunomemories, secretion of cytotoxic materials, and direct attacks on the plasma membranes of foreign cells.[11][14]
Cytotoxicity
In addition to being able to destroy infectious organisms through cell to cell contact via phagocytosis, microglia can also release a variety of cytotoxic substances. Microglia in culture secrete large amounts of H2O2 and NO in a process known as ‘respiratory burst'. Both of these chemicals can directly damage cells and lead to neuronal cell death. Proteases secreted by microglia catabolise specific proteins causing direct cellular damage, while cytokines like IL-1 promote demyelination of neuronal axons. Finally, microglia can injure neurons through NMDA receptor-mediated processes by secreting glutamate, aspartate and quinolinic acid. Cytotoxic secretion is aimed at destroying infected neurons, virus, and bacteria, but can also cause large amounts of collateral neural damage. As a result, chronic inflammatory response can result in large scale neural damage as the microglia ravage the brain in an attempt to destroy the invading infection.[11]
Synaptic stripping
In a phenomenon first noticed in spinal lesions by Blinzinger and Kreutzberg in 1968, post-inflammation microglia remove the branches from nerves near damaged tissue. This helps promote regrowth and remapping of damaged neural circuitry.[11]
Promotion of repair
Post-inflammation, microglia undergo several steps to promote regrowth of neural tissue. These include synaptic stripping, secretion of anti-inflammatory cytokines, recruitment of neurons and astrocytes to the damaged area, and formation of gitter cells. Without microglial cells regrowth and remapping would be considerably slower in the resident areas of the CNS and almost impossible in many of the vascular systems surrounding the brain and eyes.[11][13]
Role in chronic neuroinflammation
The word neuroinflammation has come to stand for chronic, central nervous system (CNS) specific, inflammation-like glial responses that may produce neurodegenerative symptoms such as plaque formation, dystrophic neurite growth, and excessive tau phosphorylation.[26] It is important to distinguish between acute and chronic neuroinflammation. Acute neuroinflammation is generally caused by some neuronal injury after which microglia migrate to the injured site engulfing dead cells and debris.[26] The term neuroinflammation generally refers to more chronic, sustained injury when the responses of microglial cells contribute to and expand the neurodestructive effects, worsening the disease process.[26]
When microglia are activated they take on an amoeboid shape and they alter their gene expression. Altered gene expression leads to the production of numerous potentially neurotoxic mediators. These mediators are important in the normal functions of microglia and their production is usually decreased once their task is complete.[27] In chronic neuroinflammation, microglia remain activated for an extended period during which the production of mediators is sustained longer than usual.[27] This increase in mediators contributes to neuronal death.[27]
Neuroinflammation is distinct from inflammation in other organs, but does include some similar mechanisms such as the localized production of chemoattractant molecules to the site of inflammation.[27] The following list contains a few of the numerous substances that are secreted when microglia are activated:
Cytokines
Microglia activate the proinflammatory cytokines IFN-γ, IL-1α, IL-1β and TNF-α in the CNS.[2][27] Direct injection of the cytokines IL-1α, IL-1β and TNF-α into the CNS result in local inflammatory responses and neuronal degradation. Cytokines play a potential role in neurodegeneration when microglia remain in a sustained activated state.[27] This is in contrast with the potential neurotrophic (inducing growth of neurons) actions of these cytokines during acute neuroinflammation.[27]
Chemokines
Chemokines are cytokines that stimulate directional migration of inflammatory cells in vitro and in vivo.[27] Chemokines are divided into four main subfamilies: C, CC, CXC, and CX3C. Microglial cells are sources of some chemokines and express the monocyte chemoattractant protein-1 (MCP-1) chemokine in particular.[27] Other inflammatory cytokines like IL-1β and TNF-α, as well as bacterial-derived lipopolysaccharide (LPS) may stimulate microglia to produce MCP-1, MIP-1α, and MIP-1β.[27] Microglia can express CCR3, CCR5, CXCL8, CXCR4, and CX3CR1 in vitro.[2][27] Chemokines are proinflammatory and therefore contribute to the neuroinflammation process.[27]
Proteases
When microglia are activated they induce the synthesis and secretion of proteolytic enzymes that are potentially involved in many functions.[27] There are a number of proteases that possess the potential to degrade both the extracellular matrix and neuronal cells that are in the neighborhood of the microglia releasing these compounds.[27] These proteases include; cathepsins B, L, and S, the matrix metalloproteinases MMP-1, MMP-2, MMP-3, and MMP-9, and the metalloprotease-disintegrin ADAM8 (plasminogen) which forms outside microglia and degrades the extracellular matrix.[27] Both Cathepsin B, MMP-1 and MMP-3 have been found to be increased in Alzheimer's disease (AD) and cathepsin B is increased in multiple sclerosis (MS).[27] Elastase, another protease, could have large negative effects on the extracellular matrix.[27]
Amyloid precursor protein
Microglia synthesize amyloid precursor protein (APP) in response to excitotoxic injury.[27] Plaques result from abnormal proteolytic cleavage of membrane bound APP.[27] Amyloid plaques can stimulate microglia to produce neurotoxic compounds such as cytokines, excitotoxin, nitric oxide and lipophylic amines, which all cause neural damage.[28] Plaques in Alzheimer's disease contain activated microglia.[27] A study has shown that direct injection of amyloid into brain tissue activates microglia, which reduces the number of neurons.[28] Microglia have also been suggested as a possible source of secreted β amyloid.[27]
Aging
Microglia undergo a burst of mitotic activity during injury; this proliferation is followed by apoptosis to reduce the cell numbers back to baseline.[26] Activation of microglia places a load on the anabolic and catabolic machinery of the cells causing activated microglia to die sooner than non-activated cells.[26] To compensate for microglial loss over time, microglia undergo mitosis and bone marrow derived progenitor cells migrate into the brain via the meninges and vasculature.[26]
Accumulation of minor neuronal damage that occurs during normal aging can transform microglia into enlarged and activated cells.[29] These chronic, age-associated increases in microglial activation and IL-1 expression may contribute to increased risk of Alzheimer's disease with advancing age through favoring neuritic plaque formation in susceptible patients.[29] DNA damage might contribute to age-associated microglial activation. Another factor might be the accumulation of advanced glycation endproducts, which accumulate with aging.[29] These proteins are strongly resistant to proteolytic processes and promote protein cross-linking.[29]
Research has discovered dystrophic (defective development) human microglia. "These cells are characterized by abnormalities in their cytoplasmic structure, such as deramified, atrophic, fragmented or unusually tortuous processes, frequently bearing spheroidal or bulbous swellings."[26] The incidence of dystrophic microglia increases with aging.[26] Microglial degeneration and death have been reported in research on Prion disease, Schizophrenia and Alzheimer's disease, indicating that microglial deterioration might be involved in neurodegenerative diseases.[26] A complication of this theory is the fact that it is difficult to distinguish between "activated" and "dystrophic" microglia in the human brain.[26]
Role in neurodegeneration
Microglia also have a role in neurodegenerative disorders, which are characterized by progressive cell loss in specific neuronal populations.[27] "Many of the normal trophic functions of glia may be lost or overwhelmed when the cells become chronically activated in progressive neurodegenerative disorders, for there is abundant evidence that in such disorders, activated glia play destructive roles by direct and indirect inflammatory attack."[27] The following are prominent examples of microglial cells' role in neurodegenerative disorders.
Alzheimer's disease
Alzheimer's disease (AD) is a progressive, neurodegenerative disease where the brain develops abnormal clumps (amyloid plaques) and tangled fiber bundles (neurofibrillary tangles).[30]
There are many activated microglia over-expressing IL-1 in the brains of Alzheimer patients that are distributed with both Aβ plaques and neurofibrillary tangles.[29] This over expression of IL-1 leads to excessive tau phosphorylation that is related to tangle development in Alzheimer's disease.[29]
Many activated microglia are found to be associated with amyloid deposits in the brains of Alzheimer's patients.[27] Microglia interact with β-amyloid plaques through cell surface receptors that are linked to tyrosine kinase based signaling cascades that induce inflammation.[27] When microglia interact with the deposited fibrillar forms of β-amyloid it leads to the conversion of the microglia into an activated cell and results in the synthesis and secretion of cytokines and other proteins that are neurotoxic.[27]
One preliminary model as to how this would occur involves a positive feedback loop. When activated, microglia will secrete proteases, cytokines, and reactive oxygen species. The cytokines may induce neighboring cells to synthesize amyloid precursor protein. The proteases then possibly could cause the cleaving required to turn precursor molecules into the beta amyloid that characterizes the disease. Then, the oxygen species encourage the aggregation of beta amyloid in order to form plaques. The growing size of these plaques then in turn triggers the action of even more microglia, which then secrete more cytokines, proteases, and oxygen species, thus amplifying the neurodegeneration.[31]
Treatment
Non-steroidal anti-inflammatory drugs (NSAIDs) have proven to be effective in reducing the risk of AD.[27] "Sustained treatment with NSAIDs lowers the risk of AD by 55%, delays disease onset, attenuates symptomatic severity and slows the loss of cognitive abilities. The main cellular target for NSAIDs is thought to be microglia. This is supported by the fact that in patients taking NSAIDs the number of activated microglia is decreased by 65%."[27]
Parkinson's disease
Parkinson's disease is a movement disorder in which the dopamine-producing neurons in the brain do not function as they should, the neurons of the Substantia Nigra become dysfunctional and eventually die, leaving a lack of dopamine input into the striatum. This causes the symptoms of Parkinson's disease.[32]
Cardiovascular Diseases
Recently microglial activation has been reported in rats with myocardial infarction (Rana et al.,2010). This activation was specific to brain nuclei involved in cardiovascular regulation suggesting possible role of microglial activation in progression to heart failure.
Role in glaucoma
Several studies have proved the changes occurring in the microglia of the inner plexiform and of the outer plexiform layers of the retina.[33][34][35] Also a new software to automatize the count of microglial cells in the retina has been published.[36]
Role in viral infections
Human immunodeficiency virus
The infection of mononuclear phagocytes with HIV-1 is an important element in the development of HIV-associated dementia complex (HAD).[37] The only brain cell type that is "productively" infected with the virus are microglial cells.[37] It has also become clear that neurotoxic mediators released from brain microglia play an important role in the pathogenesis of HIV-1.[37]
"HIV-1 can enter the microglial cell via CD4 receptors and chemokine co-receptors such as CCR3, CCR5, and CXCR4, with CCR5 being the most important of these. Interestingly, humans with double allelic loss of CCR5 are virtually immune to HIV acquired via the sexual route (though can be infected by IV transmission of CXCR4 tropic viruses). IL-4 and IL-10 enhance the entry and replication of HIV-1 in microglia through the up-regulation of CD4 and CCR5 expression, respectively. The chemokines CCL5/RANTES, CCL3/MIP-1α, CCL4/MIP-1β, all of which bind to CCR5, are inhibitory to HIV-1 replication in microglial cells, apparently by their ability to block viral entry."[37]
Infected microglia contain viral particles intracellularly.[37] There is a correlation between the severity of dementia and microglial production of neurotoxins.[37]
One discrepancy in HAD is the limited number of HIV-1 infected microglia in comparison to the many CNS abnormalities that occur.[37] This suggests that chemical factors that are released from microglial cells are contributing to neuronal loss. "It has become more and more apparent that HIV-1 infected microglial cells actively secrete both endogenous neurotoxins such as TNF-α, IL-1β, CXCL8/IL-8, glutamate, quinolinic acid, platelet activating factor, eicosanoids, and NO as well as the neurotoxic viral proteins Tat, gp120, and gp41."[37]
Microglia are the main target of HIV-1 in the brain. When activated by HIV-1 or viral proteins, they secrete or induce other cells to secrete neurotoxic factors; this process is accompanied by neuronal dysfunction (HAD).[37]
Herpes simplex virus
Herpes simplex virus (HSV) can cause herpes encephalitis in babies and immunocompetent adults. Studies have shown that long-term neuroimmune activation persists after the herpes infection in patients.[37] Microglia produce cytokines that are toxic to neurons; this may be a mechanism underlying HSV-related CNS damage.[37] It has been found that "active microglial cells in HSV encephalitis patients do persist for more than 12 months after antiviral treatment."[37]
Role in bacterial infections
Lipopolysaccharide (LPS) is the major component of the outer membrane of a gram-negative bacterial cell wall. LPS has been shown to activate microglia in vitro and stimulates microglia to produce cytokines, chemokines, and prostaglandins.[37] "Although LPS has been used as a classic activating agent, a recent study of rat microglia demonstrated that prolonged LPS exposure induces a distinctly different activated state from that in microglia acutely exposed to LPS."[37]
Streptococcus pneumoniae
Streptococcus pneumoniae is the most common cause of bacterial meningitis. It is primarily localized to the subarachnoid space while cytokines and chemokines are produced inside the blood–brain barrier.[37] Microglia interact with streptococcus via their TLR2 receptor; this interaction then activates microglia to produce nitric oxide which is neurotoxic.[38] The inflammatory response, triggered by microglia, may cause intracerebral edema.[37]
Role in parasitic infections
Plasmodium falciparum
Plasmodium falciparum is a parasite that causes malaria in humans.[37] A serious complication of malaria is cerebral malaria (CM).[37] CM occurs when red blood cells break through the blood–brain barrier, causing microhemorrhages, ischemia and glial cell growth.[37] This can lead to microglial aggregates called Durck's granulomas.[37] Recent research has indicated that microglia play a major role in the pathogenesis of CM.[37]
Role in neuropathic pain
Microglia have been implicated in neuropathic pain. They become activated in response to nerve injury, as demonstrated by several animal models.[39] Activated microglia release substances that excite pain-sensitive neurons, including prostaglandins and reactive oxygen species, through the purinergic signaling mechanisms.[40][41] Moreover, microglia also release of proinflammatory molecules through the stimulation of purinergic receptors, including IL1-β, IL-6, and TNF-α.[42][43][44] The release of these molecules is mediated by the P2X7 receptor and creates a positive feedback loop, exacerbating the pain response.[45]
A causal role for microglia in the pathogenesis of neuropathic pain has been demonstrated through P2X4 receptor.[46] P2X4 receptors are upregulated following injury and the increase in purinergic signaling activates p38-mitogen-activated protein kinase (p38 MAPK). The increase in p38 MAPK signaling leads to greater microglial release of brain-derived neurotrophic factor (BDNF).[47][48] BDNF released from microglia induces neuronal hyperexcitability through interaction with the TrkB receptor.[49]
Therapeutic development has focused on finding purinergic signaling blockers. There has been some success with P2X7 blockers, A-438079 and A-740003, however there are no selective P2X4 receptor antagonists to date.[50][51][52]
As a target to treat neuroinflammation
Inhibition of activation
One way to control neuroinflammation is to inhibit microglial activation. Studies on microglia have shown that they are activated by diverse stimuli but they are dependent on activation of mitogen-activated protein kinase (MAPK).[27] Previous approaches to down-regulate activated microglia focused on immunosuppressants.[27] Recently, minocycline (a tetracycline derivative) has shown down-regulation of microglial MAPK.[27] Another promising treatment is CPI-1189, which induces cell death in a TNF α-inhibiting compound that also down-regulates MAPK.[27] Recent study shows that nicergoline (Sermion) suppresses the production of proinflammatory cytokines and superoxide anion by activated microglia.[53] Microglial activation can be inhibited by MIF (microglia/macrophage inhibitory factor, tuftsin fragment 1–3, Thr-Lys-Pro). MIF-treated mice showed reduced brain injury and improved neurologic function in a mouse model of collagenase-induced intracerebral hemorrhage.[54][55]
Regulation of chemokine receptor
The chemokine receptor, CX3CR1, is expressed by microglia in the central nervous system.[56] Fractalkine (CX3CL1) is the exclusive ligand for CX3CR1 and is made as a transmembrane glycoprotein from which a chemokine can be released.[56] Cardona, et al. stated in 2006 that "using three different in vivo models, we show that CX3CR1 deficiency dysregulates microglial responses, resulting in neurotoxicity."[56] Further studies into how CX3CR1 regulates microglial neurotoxicity could lead to new therapeutic strategies for neuroprotection.[56]
Inhibition of amyloid deposition
Inhibitors of amyloid deposition include the enzymes responsible for the production of extracellular amyloid such as β-secretase and γ-secretase inhibitors.[27] Currently the γ-secretase inhibitors are in phase II clinical trials as a treatment for Alzheimer's disease but they have immunosuppressive properties, which could limit their use.[27] Another strategy involves increasing the antibodies against a fragment of amyloid.[27] This treatment is also in phase II clinical trials for the treatment of Alzheimer's disease.[27]
Inhibition of cytokine synthesis
Glucocorticosteroids (GCS) are anti-inflammatory steroids that inhibit both central and peripheral cytokine synthesis and action.[27] In a study conducted by Kalipada Pahan from the Department of Pediatrics at the Medical University of South Carolina, both lovastatin and sodium phenylacetate were found to inhibit TNF-α, IL-1β, and IL-6 in rat microglia.[57] This shows that the mevalonate pathway plays a role in controlling the expression of cytokines in microglia and may be important in developing drugs to treat neurodegenerative diseases.[57] Naltrexone may provide a solution to the inflammatory mediators produced by microglia. Although naltrexone's main action is to competitively bind to opioid receptors, new research shows that naltrexone, when given in low doses once per day (low-dose naltrexone), can inhibit cytokine synthesis by microglia cells. This mechanism is still being investigated, but there are already studies that indicate that it helps some patients suffering from fibromyalgia syndrome. Naltrexone shows more promise than GCSs because the GCSs inhibit immune system function more generally, increase allergic reactions and, as the name implies, increase blood glucose levels.[58][59]
See also
References
- ↑ Prinz, Marco; Tay, Tuan Leng; Wolf, Yochai; Jung, Steffen (2014). "Microglia: Unique and common features with other tissue macrophages". Acta Neuropathologica. 128 (3): 319–31. doi:10.1007/s00401-014-1267-1. PMID 24652058.
- 1 2 3 4 Shattuck, Eric C.; Muehlenbein, Michael P. (2015). "Human sickness behavior: Ultimate and proximate explanations". American Journal of Physical Anthropology. 157 (1): 1–18. doi:10.1002/ajpa.22698. PMID 25639499.
- ↑ Alboni, Silvia; Cervia, Davide; Sugama, Shuei; Conti, Bruno (2010). "Interleukin 18 in the CNS". Journal of Neuroinflammation. 7: 9. doi:10.1186/1742-2094-7-9. PMC 2830964
 . PMID 20113500.
. PMID 20113500. - ↑ Colton, C. A.; Abel, C.; Patchett, J.; Keri, J.; Yao, J. (1992). "Lectin staining of cultured CNS microglia". Journal of Histochemistry & Cytochemistry. 40 (4): 505–12. doi:10.1177/40.4.1372634. PMID 1372634.
- ↑ Frick, Luciana Romina; Williams, Kyle; Pittenger, Christopher (2013). "Microglial Dysregulation in Psychiatric Disease". Clinical and Developmental Immunology. 2013: 608654. doi:10.1155/2013/608654. PMC 3652125. PMID 23690824.
- ↑ Ginhoux, Florent; Lim, Shawn; Hoeffel, Guillaume; Low, Donovan; Huber, Tara (2013). "Origin and differentiation of microglia". Frontiers in Cellular Neuroscience. 7. doi:10.3389/fncel.2013.00045.
- ↑ Lawson, L.J.; Perry, V.H.; Gordon, S. (1992). "Turnover of resident microglia in the normal adult mouse brain". Neuroscience. 48 (2): 405–15. doi:10.1016/0306-4522(92)90500-2. PMID 1603325.
- ↑ Filiano, Anthony J.; Gadani, Sachin P.; Kipnis, Jonathan (2015). "Interactions of innate and adaptive immunity in brain development and function". Brain Research. 1617: 18–27. doi:10.1016/j.brainres.2014.07.050. PMC 4320678. PMID 25110235.
- ↑ Kreutzberg, G. W. (1995). "Microglia, the first line of defence in brain pathologies". Arzneimittel-Forschung. 45 (3A): 357–60. PMID 7763326.
- ↑ Bushong, Eric A.; Martone, Maryann E.; Jones, Ying Z.; Ellisman, Mark H. (January 2002). "Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains". The Journal of Neuroscience. 22 (1): 183–92. PMID 11756501.
- 1 2 3 4 5 6 7 8 9 10 11 12 Gehrmann, Jochen; Matsumoto, Yoh; Kreutzberg, Georg W. (1995). "Microglia: Intrinsic immuneffector cell of the brain". Brain Research Reviews. 20 (3): 269–87. doi:10.1016/0165-0173(94)00015-H. PMID 7550361.
- ↑ Dissing-Olesen, L.; Ladeby, R.; Nielsen, H.H.; Toft-Hansen, H.; Dalmau, I.; Finsen, B. (2007). "Axonal lesion-induced microglial proliferation and microglial cluster formation in the mouse". Neuroscience. 149 (1): 112–22. doi:10.1016/j.neuroscience.2007.06.037. PMID 17870248.
- 1 2 3 Ritter, Matthew R.; Banin, Eyal; Moreno, Stacey K.; Aguilar, Edith; Dorrell, Michael I.; Friedlander, Martin (2006). "Myeloid progenitors differentiate into microglia and promote vascular repair in a model of ischemic retinopathy". Journal of Clinical Investigation. 116 (12): 3266–76. doi:10.1172/JCI29683. PMC 1636693. PMID 17111048.
- 1 2 3 4 5 6 7 Aloisi, Francesca (2001). "Immune function of microglia". Glia. 36 (2): 165–79. doi:10.1002/glia.1106. PMID 11596125.
- ↑ Gehrmann, J. (1996). "Microglia: A sensor to threats in the nervous system?". Research in Virology. 147 (2–3): 79–88. doi:10.1016/0923-2516(96)80220-2. PMID 8901425.
- ↑ Babeş, VM (1892). "Certains caractères des lesions histologiques de la rage" [Certain characteristics of the histological lesions of rabies]. Annales de l'Institut Pasteur (in French). 6: 209–23.
- ↑ del Río Hortega, Pío; Penfield, Wilder (1892). "Cerebral Cicatrix: the Reaction of Neuroglia and Microglia to Brain Wounds". Bulletin of the Johns Hopkins Hospital. 41: 278–303.
- ↑ del Rio-Hortega, P (1937). "Microglia". Cytology and Cellular Pathology of the Nervous System: 481–534.
- 1 2 3 Verkhratsky, Alexei; Butt, Arthur (2013). Glial physiology and pathophysiology. Chicester: John Wiley & Sons. ISBN 1118402057.
- 1 2 3 Christensen, Randolph N.; Ha, Byeong Keun; Sun, Fang; Bresnahan, Jacqueline C.; Beattie, Michael S. (2006). "Kainate induces rapid redistribution of the actin cytoskeleton in ameboid microglia". Journal of Neuroscience Research. 84 (1): 170–81. doi:10.1002/jnr.20865. PMID 16625662.
- ↑ Davis, E.J.; Foster, T.D.; Thomas, W.E. (1994). "Cellular forms and functions of brain microglia". Brain Research Bulletin. 34 (1): 73–8. doi:10.1016/0361-9230(94)90189-9. PMID 8193937.
- ↑ Eggen, B. J. L.; Raj, D.; Hanisch, U.-K.; Boddeke, H. W. G. M. (2013). "Microglial Phenotype and Adaptation". Journal of Neuroimmune Pharmacology. 8 (4): 807–23. doi:10.1007/s11481-013-9490-4. PMID 23881706.
- ↑ Jelinek, HF; Karperien, A; Bossomaier, T; Buchan, A (1975). "Differentiating grades of microglia activation with fractal analysis" (PDF). Complexity International. 12 (18): 1713–7.
- 1 2 Ferrer, I.; Bernet, E.; Soriano, E.; Del Rio, T.; Fonseca, M. (1990). "Naturally occurring cell death in the cerebral cortex of the rat and removal of dead cells by transitory phagocytes". Neuroscience. 39 (2): 451–8. doi:10.1016/0306-4522(90)90281-8. PMID 2087266.
- ↑ Rissi, Daniel R.; Oliveira, Fabiano N.; Rech, Raquel R.; Pierezan, Felipe; Lemos, Ricardo A.A.; Barros, Claudio S.L. (2006). "Epidemiologia, sinais clínicos e distribuição das lesões encefálicas em bovinos afetados por meningoencefalite por herpesvírus bovino-5" [Epidemiology, clinical signs and distribution of the encephalic lesions in cattle affected by meningoencephalitis caused by bovine herpesvirus-5]. Pesquisa Veterinária Brasileira (in Portuguese). 26 (2): 123–32. doi:10.1590/S0100-736X2006000200010.
- 1 2 3 4 5 6 7 8 9 10 Streit, Wolfgang J. (2006). "Microglial senescence: Does the brain's immune system have an expiration date?". Trends in Neurosciences. 29 (9): 506–10. doi:10.1016/j.tins.2006.07.001. PMID 16859761.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 Wood, Paul (2003). Neuroinflammation: Mechanisms and Management. Humana Press.
- 1 2 Golden, Nyoman; Darmadipura, Sajid (2007). "The Role of Microglia as Prime Component of CNS Immune System in Acute and Chronic Neuroinflammation" (PDF). Folica Medica Indonesiana. 43 (1): 54–8.
- 1 2 3 4 5 6 Mrak, Robert E.; Griffin, W. Sue T. (2005). "Glia and their cytokines in progression of neurodegeneration". Neurobiology of Aging. 26 (3): 349–54. doi:10.1016/j.neurobiolaging.2004.05.010. PMID 15639313.
- ↑ "National Institute of Neurological Disorders and Stroke". NINDS Alzheimer's Disease Information Page. November 14, 2007.
- ↑ Streit, Wolfgang J.; Kincaid-Colton, Carol A. (1995). "The Brain's Immune System". Scientific American. 273 (5): 54–5, 58–61. doi:10.1038/scientificamerican1195-54. PMID 8966536.
- ↑ "Parkinson's Disease: Hope Through Research". National Institute of Neurological Disorders and Stroke. November 13, 2007.
- ↑ Rojas, Blanca; Gallego, Beatriz I; Ramírez, Ana I; Salazar, Juan J; De Hoz, Rosa; Valiente-Soriano, Francisco J; Avilés-Trigueros, Marcelino; Villegas-Perez, Maria P; Vidal-Sanz, Manuel; Triviño, Alberto; Ramírez, José M (2014). "Microglia in mouse retina contralateral to experimental glaucoma exhibit multiple signs of activation in all retinal layers". Journal of Neuroinflammation. 11: 133. doi:10.1186/1742-2094-11-133. PMC 4128533. PMID 25064005.
- ↑ De Hoz, Rosa; Gallego, Beatriz I.; Ramírez, Ana I.; Rojas, Blanca; Salazar, Juan J.; Valiente-Soriano, Francisco J.; Avilés-Trigueros, Marcelino; Villegas-Perez, Maria P.; Vidal-Sanz, Manuel; Triviño, Alberto; Ramírez, José M. (2013). "Rod-Like Microglia Are Restricted to Eyes with Laser-Induced Ocular Hypertension but Absent from the Microglial Changes in the Contralateral Untreated Eye". PLoS ONE. 8 (12): e83733. Bibcode:2013PLoSO...883733D. doi:10.1371/journal.pone.0083733. PMID 24367610.
- ↑ Gallego, Beatriz I; Salazar, Juan J; De Hoz, Rosa; Rojas, Blanca; Ramírez, Ana I; Salinas-Navarro, Manuel; Ortín-Martínez, Arturo; Valiente-Soriano, Francisco J; Avilés-Trigueros, Marcelino; Villegas-Perez, Maria P; Vidal-Sanz, Manuel; Triviño, Alberto; Ramírez, Jose M (2012). "IOP induces upregulation of GFAP and MHC-II and microglia reactivity in mice retina contralateral to experimental glaucoma". Journal of Neuroinflammation. 9. doi:10.1186/1742-2094-9-92.
- ↑ De Gracia, Pablo; Gallego, Beatriz I.; Rojas, Blanca; Ramírez, Ana I.; De Hoz, Rosa; Salazar, Juan J.; Triviño, Alberto; Ramírez, José M. (2015). "Automatic Counting of Microglial Cells in Healthy and Glaucomatous Mouse Retinas". PLoS ONE. 10 (11): e0143278. Bibcode:2015PLoSO..1043278D. doi:10.1371/journal.pone.0143278. PMID 26580208.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 Rock, R. B.; Gekker, G.; Hu, S.; Sheng, W. S.; Cheeran, M.; Lokensgard, J. R.; Peterson, P. K. (2004). "Role of Microglia in Central Nervous System Infections". Clinical Microbiology Reviews. 17 (4): 942–64, table of contents. doi:10.1128/CMR.17.4.942-964.2004. PMC 523558. PMID 15489356.
- ↑ Lehnardt, S.; Wennekamp, J.; Freyer, D.; Liedtke, C.; Krueger, C.; Nitsch, R.; Bechmann, I.; Weber, J. R.; Henneke, P. (2007). "TLR2 and Caspase-8 Are Essential for Group B Streptococcus-Induced Apoptosis in Microglia". The Journal of Immunology. 179 (9): 6134–43. doi:10.4049/jimmunol.179.9.6134. PMID 17947688.
- ↑ Watkins, Linda R.; Milligan, Erin D.; Maier, Steven F. (2001). "Glial activation: A driving force for pathological pain". Trends in Neurosciences. 24 (8): 450–5. doi:10.1016/S0166-2236(00)01854-3. PMID 11476884.
- ↑ Barbera-Cremades, M.; Baroja-Mazo, A.; Gomez, A. I.; Machado, F.; Di Virgilio, F.; Pelegrin, P. (2012). "P2X7 receptor-stimulation causes fever via PGE2 and IL-1 release". The FASEB Journal. 26 (7): 2951–62. doi:10.1096/fj.12-205765. PMID 22490780.
- ↑ Bartlett, Rachael; Yerbury, Justin J.; Sluyter, Ronald (2013). "P2X7 Receptor Activation Induces Reactive Oxygen Species Formation and Cell Death in Murine EOC13 Microglia". Mediators of Inflammation. 2013: 271813. doi:10.1155/2013/271813. PMC 3568910. PMID 23431238.
- ↑ Clark, A. K.; Staniland, A. A.; Marchand, F.; Kaan, T. K. Y.; McMahon, S. B.; Malcangio, M. (2010). "P2X7-Dependent Release of Interleukin-1 and Nociception in the Spinal Cord following Lipopolysaccharide". Journal of Neuroscience. 30 (2): 573–82. doi:10.1523/JNEUROSCI.3295-09.2010. PMC 2880485. PMID 20071520.
- ↑ Shigemoto-Mogami, Yukari; Koizumi, Schuichi; Tsuda, Makoto; Ohsawa, Keiko; Kohsaka, Shinichi; Inoue, Kazuhide (2001). "Mechanisms underlying extracellular ATP-evoked interleukin-6 release in mouse microglial cell line, MG-5". Journal of Neurochemistry. 78 (6): 1339–49. doi:10.1046/j.1471-4159.2001.00514.x. PMID 11579142.
- ↑ Hide, Izumi; Tanaka, Masaya; Inoue, Atsuko; Nakajima, Kazuyuki; Kohsaka, Shinichi; Inoue, Kazuhide; Nakata, Yoshihiro (2002). "Extracellular ATP Triggers Tumor Necrosis Factor-α Release from Rat Microglia". Journal of Neurochemistry. 75 (3): 965–72. doi:10.1046/j.1471-4159.2000.0750965.x. PMID 10936177.
- ↑ Hains, B. C.; Waxman, S. G. (2006). "Activated Microglia Contribute to the Maintenance of Chronic Pain after Spinal Cord Injury". Journal of Neuroscience. 26 (16): 4308–17. doi:10.1523/JNEUROSCI.0003-06.2006. PMID 16624951.
- ↑ Tsuda, Makoto; Shigemoto-Mogami, Yukari; Koizumi, Schuichi; Mizokoshi, Akito; Kohsaka, Shinichi; Salter, Michael W.; Inoue, Kazuhide (2003). "P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury". Nature. 424 (6950): 778–83. Bibcode:2003Natur.424..778T. doi:10.1038/nature01786. PMID 12917686.
- ↑ Ulmann, L.; Hatcher, J. P.; Hughes, J. P.; Chaumont, S.; Green, P. J.; Conquet, F.; Buell, G. N.; Reeve, A. J.; Chessell, I. P.; Rassendren, F. (2008). "Up-Regulation of P2X4 Receptors in Spinal Microglia after Peripheral Nerve Injury Mediates BDNF Release and Neuropathic Pain". Journal of Neuroscience. 28 (44): 11263–8. doi:10.1523/JNEUROSCI.2308-08.2008. PMID 18971468.
- ↑ Trang, T.; Beggs, S.; Wan, X.; Salter, M. W. (2009). "P2X4-Receptor-Mediated Synthesis and Release of Brain-Derived Neurotrophic Factor in Microglia is Dependent on Calcium and p38-Mitogen-Activated Protein Kinase Activation". Journal of Neuroscience. 29 (11): 3518–28. doi:10.1523/JNEUROSCI.5714-08.2009. PMC 3589565. PMID 19295157.
- ↑ Coull, Jeffrey A. M.; Beggs, Simon; Boudreau, Dominic; Boivin, Dominick; Tsuda, Makoto; Inoue, Kazuhide; Gravel, Claude; Salter, Michael W.; De Koninck, Yves (2005). "BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain". Nature. 438 (7070): 1017–21. Bibcode:2005Natur.438.1017C. doi:10.1038/nature04223. PMID 16355225.
- ↑ Honore, P.; Donnelly-Roberts, D.; Namovic, M. T.; Hsieh, G.; Zhu, C. Z.; Mikusa, J. P.; Hernandez, G.; Zhong, C.; Gauvin, D. M.; Chandran, P.; Harris, R.; Medrano, A. P.; Carroll, W.; Marsh, K.; Sullivan, J. P.; Faltynek, C. R.; Jarvis, M. F. (2006). "A-740003 [N-(1-{[(Cyanoimino)(5-quinolinylamino) methyl]amino}-2,2-dimethylpropyl)-2-(3,4-dimethoxyphenyl)acetamide], a Novel and Selective P2X7 Receptor Antagonist, Dose-Dependently Reduces Neuropathic Pain in the Rat". Journal of Pharmacology and Experimental Therapeutics. 319 (3): 1376–85. doi:10.1124/jpet.106.111559. PMID 16982702.
- ↑ Nelson, Derek W.; Gregg, Robert J.; Kort, Michael E.; Perez-Medrano, Arturo; Voight, Eric A.; Wang, Ying; Grayson, George; Namovic, Marian T.; Donnelly-Roberts, Diana L.; Niforatos, Wende; Honore, Prisca; Jarvis, Michael F.; Faltynek, Connie R.; Carroll, William A. (2006). "Structure−Activity Relationship Studies on a Series of Novel, Substituted 1-Benzyl-5-phenyltetrazole P2X7Antagonists". Journal of Medicinal Chemistry. 49 (12): 3659–66. doi:10.1021/jm051202e. PMID 16759108.
- ↑ Burnstock, Geoffrey (2013). "Purinergic mechanisms and pain—An update". European Journal of Pharmacology. 716 (1–3): 24–40. doi:10.1016/j.ejphar.2013.01.078. PMID 23524093.
- ↑ Mizuno, Tetsuya; Kuno, Reiko; Nitta, Atsumi; Nabeshima, Toshitaka; Zhang, Guiqin; Kawanokuchi, Jun; Wang, Jinyan; Jin, Shijie; Takeuchi, Hideyuki; Suzumura, Akio (2005). "Protective effects of nicergoline against neuronal cell death induced by activated microglia and astrocytes". Brain Research. 1066 (1–2): 78–85. doi:10.1016/j.brainres.2005.10.050. PMID 16325157.
- ↑ Wang, Jian; Rogove, Andrew D.; Tsirka, Anna E.; Tsirka, Stella E. (2003). "Protective role of tuftsin fragment 1-3 in an animal model of intracerebral hemorrhage". Annals of Neurology. 54 (5): 655–64. doi:10.1002/ana.10750. PMID 14595655.
- ↑ Wang, J.; Tsirka, S. E. (2005). "Tuftsin Fragment 1-3 is Beneficial when Delivered After the Induction of Intracerebral Hemorrhage". Stroke. 36 (3): 613–8. doi:10.1161/01.STR.0000155729.12931.8f. PMID 15692122.
- 1 2 3 4 Cardona, Astrid E; Pioro, Erik P; Sasse, Margaret E; Kostenko, Volodymyr; Cardona, Sandra M; Dijkstra, Ineke M; Huang, Deren; Kidd, Grahame; Dombrowski, Stephen; Dutta, Ranjan; Lee, Jar-Chi; Cook, Donald N; Jung, Steffen; Lira, Sergio A; Littman, Dan R; Ransohoff, Richard M (2006). "Control of microglial neurotoxicity by the fractalkine receptor". Nature Neuroscience. 9 (7): 917–24. doi:10.1038/nn1715. PMID 16732273.
- 1 2 Pahan, K; Sheikh, F G; Namboodiri, A M; Singh, I (1997). "Lovastatin and phenylacetate inhibit the induction of nitric oxide synthase and cytokines in rat primary astrocytes, microglia, and macrophages". Journal of Clinical Investigation. 100 (11): 2671–9. doi:10.1172/JCI119812. PMC 508470. PMID 9389730.
- ↑ Younger, Jarred; MacKey, Sean (2009). "Fibromyalgia Symptoms Are Reduced by Low-Dose Naltrexone: A Pilot Study". Pain Medicine. 10 (4): 663–72. doi:10.1111/j.1526-4637.2009.00613.x. PMC 2891387. PMID 19453963.
- ↑ Younger, Jarred; Noor, Noorulain; McCue, Rebecca; MacKey, Sean (2013). "Low-dose naltrexone for the treatment of fibromyalgia: Findings of a small, randomized, double-blind, placebo-controlled, counterbalanced, crossover trial assessing daily pain levels". Arthritis & Rheumatism. 65 (2): 529–38. doi:10.1002/art.37734. PMID 23359310.
External links
| Wikimedia Commons has media related to Microglia. |
- Microglia home page at microglia.net
- Rock, R. B.; Gekker, G.; Hu, S.; Sheng, W. S.; Cheeran, M.; Lokensgard, J. R.; Peterson, P. K. (2004). "Role of Microglia in Central Nervous System Infections". Clinical Microbiology Reviews. 17 (4): 942–64, table of contents. doi:10.1128/CMR.17.4.942-964.2004. PMC 523558. PMID 15489356.
- Creeping into your Head - A Brief Introduction to Microglia — A Review from the Science Creative Quarterly
- "Immune Scavengers Target Alzheimer's Plaques". April 6, 2007.
- The Department of Neuroscience at Wikiversity
- NIF Search - Microglial Cell via the Neuroscience Information Framework