Myristoylation
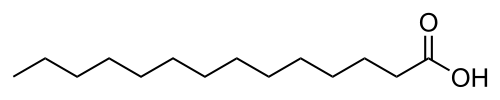
Myristoylation is a lipidation modification where a myristoyl group, derived from myristic acid, is covalently attached by an amide bond to the alpha-amino group of an N-terminal glycine residue.[1] Myristic acid is a 14-carbon saturated fatty acid (14:0) with the systematic name of n-Tetradecanoic acid. This modification can be added either co-translationally or post-translationally. N-myristoyltransferase (NMT) catalyzes the myristic acid addition reaction in the cytoplasm of cells.[2] This lipidation event is common among many organisms including animals, plants, fungi, protozoans [3] and viruses. Myristoylation allows for weak protein–protein and protein–lipid interactions[4] and plays an essential role in membrane targeting, protein–protein interactions and functions widely in a variety of signal transduction pathways.
Discovery
In 1982, Koiti Titani's lab identified an "N-terminal blocking group" on the catalytic subunit of cyclic AMP-dependent protein kinase from cow as n-Tetradecanoyl.[5] Almost simultaneously in Claude B. Klee's lab, this same N-terminal blocking group was further characterized as myristic acid.[6] Both labs made this discovery utilizing similar techniques: fast atom bombardment mass spectrometry and gas chromatography.[5][6]
N-myristoyltransferase
The enzyme N-myristoyltransferase (NMT) is responsible for the irreversible addition of a myristoyl group to N-terminal or internal glycine residues of proteins. This modification can occur co-translationally or post-translationally. In vertebrates, this modification is carried about by two NMTs, NMT1 and NMT2, both of which are members of the GCN5 acetyltransferase superfamily.[7]
Structure
The crystal structure of NMT reveals two identical subunits, each with its own myristoyl CoA binding site. Each subunit consists of a large saddle-shaped β-sheet surrounded by α-helices. The symmetry of the fold is pseudo two-fold. Myristoyl CoA binds at the N-terminal portion while the C-terminal end binds the protein.[8]
Mechanism
The addition of the myristoyl group proceeds via a nucleophilic addition-elimination reaction. First, myristoyl coenzyme A (CoA) is positioned in its binding pocket of NMT so that the carbonyl faces two amino acid residues, phenylalanine 170 and leucine 171.[8] This polarizes the carbonyl so that there is a net positive charge on the carbon making it susceptible to nucleophilic attack by the glycine residue of the protein to be modified. When myristoyl CoA binds, NMT reorients to allow binding of the peptide. The C-terminus of NMT then acts as a general base to deprotonate the NH3+ activating the amino group to attack at the carbonyl of Myristoyl CoA. The resulting tetrahedral intermediate is stabilized by the interaction between a positively charged oxyanion hole and the negatively charged alkoxide anion. Free CoA is then released resulting in a conformational change in the enzyme allowing the release of the myristoylated peptide.[2]

Co-translational vs post-translational addition
Co-translational and post-translational covalent modifications enable proteins to develop higher levels of complexity in cellular function, further adding diversity to the proteome.[9] The addition of myristoyl CoA to a protein can occur during protein translation or after. During co-translational addition of the myristoyl group, the N-terminal glycine is modified following cleavage of the N-terminal methionine residue in the newly forming, growing polypeptide.[1] This occurs in approximately 80% of myristoylated proteins. Post-translational myristoylation typically occurs following a caspase cleavage event resulting in the exposure of an internal glycine residue, which would then be available for myristic acid addition.[7]
Functions
Myristoylated proteins
| Protein | Physiological Role | Myristoylation Function |
|---|---|---|
| Actin | Cytoskeleton structural protein | Post-translational myristoylation during apoptosis [7] |
| Bid | Apoptosis promoting protein | Post-translational myristoylation after caspase cleavage targets protein to mitochondrial membrane [7] |
| MARCKS | actin cross-linking when phosphorylated by protein kinase C | Co-translational myristoylation aids in plasma membrane association |
| G-Protein | Signaling GTPase | Co-translational myristoylation aids in plasma membrane association[10] |
| Gelsolin | Actin filament-severing protein | Post-translational myristoylation up-regulates anti-apoptotic properties [7] |
| PAK2 | Serine/threonine kinase cell growth, mobility, survival stimulator | Post-translational myristoylation up-regulates apoptotic properties and induces plasma membrane localization[7] |
| Arf | vesicular trafficking and actin remodeling regulation | N-terminus myristoylation aids in membrane association |
| Hippocalcin | Neuronal calcium sensor | Contains a Ca2+/myristoyl switch |
Myristoylation molecular switch


Myristoylation not only diversifies the function of a protein, but can also add layers of regulation. One of the major, most common functions of the myristoyl group is in membrane association and cellular localization of the modified protein. Though the myristoyl group is added onto the end of the protein, in some cases it is sequestered within hydrophobic regions in the protein rather than solvent exposed.[4] By regulating the orientation of the myristoyl group on the protein, these processes can be highly coordinated and closely controlled. This defines myristoylation as a “molecular switch”.[11]
Both a hydrophobic myristoyl group and a “basic patch”, or highly positive regions on the protein characterize myristoyl-electrostatic switches. The basic patch allows for favorable electrostatic interactions to occur between the negatively charged phospholipid-heads of the membrane and the positive surface of the associating protein. This allows tighter association and directed localization of proteins.[4]
Myristoyl-conformational switches can come in several forms. Ligand binding to a myristoylated protein with its myristoyl group sequestered can result in a conformational change in the protein resulting in the exposure of the myristoyl group. Similarly, some myristoylated proteins are activated not by a designated ligand, but by the exchange of GDP for GTP by guanine nucleotide exchange factors (GEFs) in the cell. Once GTP is bound to the myristoylated protein, it becomes activated, exposing the myristoyl group. These conformational switches can be utilized as a signal for cellular localization, membrane-protein and protein–protein interactions. [4] [11] [12]
Dual modifications of myristoylated proteins
Further modifications on N-myristoylated proteins can add another level of regulation for myristoylated protein. Dual acylation of proteins can facilitate more tightly regulated protein localization, specifically targeting proteins to lipid rafts at membranes[13] or additionally allowing dissociation of myristoylated proteins from membranes.
Myristoylation and palmitoylation are commonly coupled modifications. Myristoylation alone can promote transient membrane interactions[4] that enable proteins to anchor to membranes but dissociate easily. Further palmitoylation allows for tighter anchoring and slower dissociation from membranes when required by the cell. This specific dual modification is important for GPCR pathways and is referred to as the dual fatty acylation switch.[4][7]
Myristoylation is additionally often followed by phosphorylation of nearby residues. Additional phosphorylation of the same protein can decrease the electrostatic affinity of the myristoylated protein for the membrane, causing translocation of that protein to the cytoplasm following dissociation from the membrane.[4]
Signal transduction
Myristoylation plays a vital role in membrane targeting and signal transduction[14] in plant responses to environmental stress. In addition, in signal transduction via G protein, palmitoylation of the α subunit, prenylation of the γ subunit, and myristoylation is involved in tethering the G protein to the inner surface of the plasma membrane so that the G protein can interact with its receptor.[15]
Apoptosis
Myristoylation is an integral part of apoptosis, or programmed cell death. Apoptosis is necessary for cell homeostasis and occurs when cells are under stress such as hypoxia or DNA damage. Apoptosis can proceed by either mitochondrial or receptor mediated activation. In receptor mediated apoptosis, apoptotic pathways are triggered when the cell binds a death receptor. In one such case, death receptor binding initiates the formation of the death-inducing signaling complex, a complex composed of numerous proteins including several caspases, including caspase 3. Caspase 3 cleaves a number of proteins that are subsequently myristoylated by NMT. The pro-apoptotic BH3-interacting domain death agonist (Bid) is one such protein that once myristoylated, translocates to the mitochondria where it prompts the release of cytochrome c leading to cell death.[7] Actin, gelsolin and p21-activated kinase 2 PAK2 are three other proteins that are myristoylated following cleavage by caspase 3, which leads to either the up-regulation or down-regulation of apoptosis.[7]
Impact on human health
Cancer
c-Src is a gene that is important for normal mitotic cycling. It is phosphorylated and dephosphorylated turning signaling on and off. Proto-oncogene tyrosine-protein kinase Src must be localized to the plasma membrane in order to phosphorylate other downstream targets; myristoylation is responsible for this membrane targeting event. Increased myristoylation of c-Src can lead to enhanced cell proliferation and can be responsible for transforming normal cells into cancer cells.[4][12][16] Activation of c-Src can lead to upregulation of angiogenesis, proliferation and invasion: The Hallmarks of Cancer.[17]
Viral infectivity

HIV-1 is a retrovirus that relies on myristoylation of one of its structural proteins in order to successfully package its genome, assemble and mature into a new infectious particle. Viral matrix protein, the N-terminal most domain of the gag polyprotein is myristoylated.[18] This myristoylation modification targets gag to the membrane of the host cell. Utilizing the myristoyl-electrostatic switch,[11] including a basic patch on the matrix protein, gag can assemble at lipid rafts at the plasma membrane for viral assembly, budding and further maturation.[16] In order to prevent viral infectivity, myristoylation of the matrix protein could become a good drug target.
Prokaryotic and eukaryotic infections
Certain NMTs are therapeutic targets for development of drugs against bacterial infections. Myristoylation has been shown to be necessary for the survival of a number of disease-causing fungi including C. albicans and C. neoformans. In addition to prokaryotic bacteria, the NMTs of numerous disease-causing eukaryotic organisms have been identified as drug targets as well. Proper NMT functioning in the protozoa Leishmania major and Leishmania donovani (leishmaniasis), Trypanosoma brucei (African sleeping sickness) and P. falciparum (malaria) is necessary for survival of the parasites. Inhibitors of these organisms is under current investigation. A pyrazole sulphonamide inhibitor has been identified that selectively binds T. brucei, competing for the peptide binding site, thus inhibiting enzymatic activity and eliminating the parasite from the bloodstream of mice with African sleeping sickness.[7]
See also
References
- 1 2 Cox, David L. Nelson, Michael M. (2005). Lehninger principles of biochemistry (4th ed.). New York: W.H. Freeman. ISBN 0716743396.
- 1 2 Tamanoi, edited by Fuyuhiko; Sigman, David S. (2001). Protein lipidation (3rd ed.). San Diego, CA: Academic Press. ISBN 978-0-12-122722-7.
- ↑ Kara, UA; Stenzel, DJ; Ingram, LT; Bushell, GR; Lopez, JA; Kidson, C (Apr 1988). "Inhibitory monoclonal antibody against a (myristylated) small-molecular-weight antigen from Plasmodium falciparum associated with the parasitophorous vacuole membrane.". Infection and immunity. 56 (4): 903–9. PMC 259388
 . PMID 3278984.
. PMID 3278984. - 1 2 3 4 5 6 7 8 Farazi, T. A. (29 August 2001). "The Biology and Enzymology of Protein N-Myristoylation". Journal of Biological Chemistry. 276 (43): 39501–39504. doi:10.1074/jbc.R100042200.
- 1 2 Carr, SA; Biemann, K; Shoji, S; Parmelee, DC; Titani, K (Oct 1982). "n-Tetradecanoyl is the NH2-terminal blocking group of the catalytic subunit of cyclic AMP-dependent protein kinase from bovine cardiac muscle.". Proceedings of the National Academy of Sciences of the United States of America. 79 (20): 6128–31. doi:10.1073/pnas.79.20.6128. PMC 347072. PMID 6959104.
- 1 2 Aitken, A; Cohen, P; Santikarn, S; Williams, DH; Calder, AG; Smith, A; Klee, CB (Dec 27, 1982). "Identification of the NH2-terminal blocking group of calcineurin B as myristic acid.". FEBS Letters. 150 (2): 314–8. doi:10.1016/0014-5793(82)80759-x. PMID 7160476.
- 1 2 3 4 5 6 7 8 9 10 Martin, Dale D.O.; Beauchamp, Erwan; Berthiaume, Luc G. (January 2011). "Post-translational myristoylation: Fat matters in cellular life and death". Biochimie. 93 (1): 18–31. doi:10.1016/j.biochi.2010.10.018.
- 1 2 Bhatnagar, RS; Fütterer, K; Waksman, G; Gordon, JI (Nov 23, 1999). "The structure of myristoyl-CoA:protein N-myristoyltransferase.". Biochimica et Biophysica Acta. 1441 (2-3): 162–72. doi:10.1016/s1388-1981(99)00155-9. PMID 10570244.
- ↑ Snider, Jared. "Overview of Post-Translational Modifications (PTMs)". Thermo Scientific.
- ↑ Chen, Katherine A.; Manning, David R. (2001). "Regulation of G proteins by covalent modification". Oncogene. 20: 1643–1652. doi:10.1038/sj.onc.1204185. PMID 11313912.
- 1 2 3 McLaughlin, Stuart; Aderem, Alan (July 1995). "The myristoyl-electrostatic switch: a modulator of reversible protein–membrane interactions". Trends in Biochemical Sciences. 20 (7): 272–276. doi:10.1016/S0968-0004(00)89042-8.
- 1 2 Wright, Megan H.; Heal, William P.; Mann, David J.; Tate, Edward W. (7 November 2009). "Protein myristoylation in health and disease". Journal of Chemical Biology. 3 (1): 19–35. doi:10.1007/s12154-009-0032-8.
- ↑ Levental, Ilya; Grzybek, Michal; Simons, Kai (3 August 2010). "Greasing Their Way: Lipid Modifications Determine Protein Association with Membrane Rafts". Biochemistry. 49 (30): 6305–6316. doi:10.1021/bi100882y.
- ↑ HAYASHI, Nobuhiro; TITANI, Koiti (2010). "N-myristoylated proteins, key components in intracellular signal transduction systems enabling rapid and flexible cell responses". Proceedings of the Japan Academy, Series B. 86 (5): 494–508. doi:10.2183/pjab.86.494.
- ↑ Wall, Mark A.; Coleman, David E.; Lee, Ethan; Iñiguez-Lluhi, Jorge A.; Posner, Bruce A.; Gilman, Alfred G.; Sprang, Stephen R. (December 1995). "The structure of the G protein heterotrimer Giα1β1γ2". Cell. 83 (6): 1047–1058. doi:10.1016/0092-8674(95)90220-1. PMID 8521505.
- 1 2 Shoji, S; Kubota, Y (Feb 1989). "[Function of protein myristoylation in cellular regulation and viral proliferation].". Yakugaku Zasshi. 109 (2): 71–85. PMID 2545855.
- ↑ Hanahan, Douglas; Weinberg, Robert A. (March 2011). "Hallmarks of Cancer: The Next Generation". Cell. 144 (5): 646–674. doi:10.1016/j.cell.2011.02.013. PMID 21376230.
- ↑ Hearps, AC; Jans, DA (Mar 2007). "Regulating the functions of the HIV-1 matrix protein.". AIDS Research and Human Retroviruses. 23 (3): 341–6. doi:10.1089/aid.2006.0108. PMID 17411366.