Oxocarbenium

An oxocarbenium ion is a canonical form of a carbocation, real or hypothetical, which places a formal positive charge on an oxygen atom.[1] They are common reactive intermediates in the hydrolysis of glycosidic bonds, and are a commonly used strategy for chemical glycosylation. These ions have since been proposed as reactive intermediates in a wide range of chemical transformations, and have been utilized in the total synthesis of several natural products. In addition, they commonly appear in mechanisms of enzyme-catalyzed biosynthesis and hydrolysis of carbohydrates in nature.
Structure
The general structure of an oxocarbenium ion contains an oxygen–carbon double bond, with the oxygen atom attached to an additional group. The ion has a formal positive charge, which is placed on the oxygen atom. The oxocarbenium cation is stabilized by resonance, which places the positive charge on the carbon. Compared to a ketone, the oxocarbenium ion is more polar, and more reactive with nucleophiles. A common example of this is the activation of a ketone by the addition of a Lewis Acid to the oxygen. An average dipole moment for a general ketone R2CO is δ = 0.51. With the addition of an acidic hydrogen to the oxygen atom to produce [R2COH]+, the dipole moment increases to δ = 0.61, making the oxocarbenium ion more susceptible to nucleophilic addition than the ketone.
Formation
Formation of oxocarbenium ions can proceed through several different pathways. Most commonly, the oxygen of a ketone will bind to a Lewis Acid, which activates the ketone, making it a more effective electrophile. The Lewis acid can be a wide range of molecules, from a simple hydrogen atom to metal complexes. The remainder of this article will focus on alkyl oxocarbenium ions, however, where the atom added to the oxygen is a carbon. One way that this sort of ion will form is the elimination of a leaving group. In carbohydrate chemistry, this leaving group is often an ether or ester. An alternative to elimination is direct deprotonation of the molecule to form the ion, however, this can be difficult and require strong bases to achieve.

Applications to synthesis
5-membered rings

A model for predicting the stereochemistry involved in the reactions of five-membered rings was proposed by Woerpel in 1999. By looking at the C-glycosylation of ribose, they determined that the stereochemistry present at C3 was driving the stereochemistry of the allyl addition. This led them to the development of the envelope transition state model to explain these effects. A nucleophile favors addition from the "inside" of the envelope, or from the top of the figure on the right. The "inside" addition produces a results in a staggered conformation, rather than the eclipsed conformation that results from the "outside" addition. By determining the lowest energy conformation based on the substituents of the ring, one can assume an "inside" addition can correct the stereochemical outcome.[2]

6-membered rings

The transition state model for a six-membered oxocarbenium ring was proposed earlier in 1992 by Woods et al.[3] The general strategy for determining the stereochemistry of a nucleophilic addition to a six-membered ring follows a similar procedure to the case of the five-membered ring. The assumption that one makes for this analysis is that the ring is in the same conformation as cyclohexene, with three carbons and the oxygen in a plane with the two other carbon atome puckered out of the plane, with one above and one below (see the figure to the right). Based on the substituients present on the ring, the lowest energy conformation is determined, keeping in mind steric and steroelectronic effects (see the section below for a discussion of stereoelectronic effects in oxocarbenium rings). Once this conformation is established, one can consider the nucleophilic addition. The addition will proceed through the low energy chair transition state, rather than the relatively high energy twist-boat. An example of this type of reaction can be seen below. The example also highlights how the stereoelectronic effect exerted by an electronegative substituent flips the lowest energy conformation and leads to opposite selectivity.[4]

Stereoelectronic effects
In an alkene ring that does not contain an oxygen atom, any large substituent prefers to be in an equatorial position, in order to minimize steric effects. It has been observed in rings containing oxocarbenium ions that electronegative substituents prefer the axial or pseudo-axial positions. When the electronegative atom is in the axial position, its electron density can be donated through space to the positively charged oxygen atom in the ring.[5] This electronic interaction stabilizes the axial conformation. Hydroxyl groups, ethers and halogens are examples of substituents that exhibit this phenomenon. Stereoelectronic effects must be taken into consideration when determining the lowest energy conformation in the analysis for nucleophilic addition to an oxocarbenium ion.[4]

Cycloadditions
In organic synthesis, vinyl oxocarbenium ions (structure on right) can be utilized in a wide range of cycloaddition reactions. They are commonly employed as dienophiles in the Diels–Alder reaction. An electron withdrawing ketone is often added to the dienophile to increase the rate of the reaction,[6] and these ketones are often converted to vinyl oxocarbenium ions during the reaction [7] It is not clear that an oxocarbenium ion necessarily will form, but Roush and co-workers demonstrated the oxocarbenium intermediate in the cyclization shown below. Two products were observed in this reaction, which could only form if the oxocarbenium ring is present as an intermediate.[7] [4+3], [2+2], [3+2] and [5+2] cycloadditions with oxocarbenium intermediates have also been reported [7]

Aldol reaction
Chiral oxocarbenium ions have been exploited to carry out highly diastereoselective and enantioselective acetate aldol addition reactions.[8] The oxocarbenium ion is used as an electrophile in the reaction. When the methyl group increases in size, the diastereoselevtivity increases.
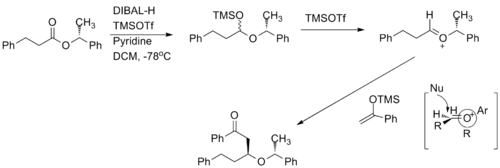
Examples from total synthesis
Oxocarbenium ions have been utilized in total synthesis on several occasions. A major subunit of (+)-clavosolide was synthesized with a reduction of a six-membered oxocarbenium ring. All the large substituents were found in an equatorial position, and the transformation went through the chair transition state, as predicted.[9]
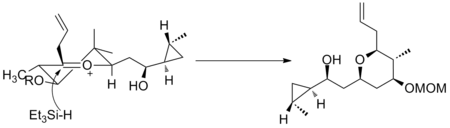
Another example is seen in the key step of the synthesis of (−)-neopeltolide, which uses another six-membered oxocarbenium ring reduction for a diastereoselective hydride addition.[10]

Applications to biology
In biological systems, oxocarbenium ions are mostly seen during reactions of carbohydrates. Since sugars are present in the structure of nucleic acids, with a ribose sugar present in RNA and a deoxyribose present in the structure of DNA, their chemistry plays an important role in wide range of cellular functions of nucleic acids. In addition to their functions in nucleotides, sugars are also used for structural components of organisms, as energy storage molecules, cell signaling molecules, protein modification and play key roles in the immune system, fertilization, preventing pathogenesis, blood clotting, and development.[11] The abundance of sugar chemistry in biological processes leads many reaction mechanisms to proceed through oxocarbenium ions. Several important biological reactions that utilize oxocarbenium ions are outlined in this section.
Nucleotide biosynthesis
Nucleotides can undergo enzyme-catalyzed intramolecular cyclization in order to produce several important biological molecules. These cyclizations typically proceed through an oxocarbenium intermediate. An example of this reaction can be seen in the cyclization cyclic ADP ribose, which is an important molecule for intracellular calcium signaling.[12]
Glycosidases
A glycosidase is an enzyme that catalyzes the breakdown of a glycosidic linkage to produce two smaller sugars. This process has important implications in the utilization of stored energy, like glycogen in animals, as well as in the breakdown of cellulose by organisms that feed on plants. In general, aspartic or glutamic acid residues in the active site of the enzyme catalyze the hydrolysis of the glycosidic bond. The mechanism of these enzymes involves an oxocarbenium ion intermediate, a general example of which is shown below.[13]

See also
References
- ↑ "IUPAC Gold Book". Retrieved 2010-12-09.
- ↑ Larsen, C. H.; Ridgway, B. H.; Shaw, J. T.; Woerpel, K. A. (1999), "A Stereoelectronic Model to Explain the Highly Stereoselective Reactions of Nucleophiles with Five-Membered-Ring Oxocarbenium Ions", Journal of the American Chemical Society, 121 (51): 12208–12209, doi:10.1021/ja993349z
- ↑ Woods, R. J.; Andrews, C. W.; Bowen, J. P. (1992), "Molecular mechanical investigations of the properties of oxocarbenium ions. 2. Application to glycoside hydrolysis", Journal of the American Chemical Society, 114 (3): 859–864, doi:10.1021/ja00029a008
- 1 2 Romero, J. A. C.; Tabacco, S. A.; Woerpel, K. A. (1999), "Stereochemical Reversal of Nucleophilic Substitution Reactions Depending upon Substituent: Reactions of Heteroatom-Substituted Six-Membered-Ring Oxocarbenium Ions through Pseudoaxial Conformers", Journal of the American Chemical Society, 122: 168–169, doi:10.1021/ja993366o
- ↑ Miljkovic, M. i.; Yeagley, D.; Deslongchamps, P.; Dory, Y. L. (1997), "Experimental and Theoretical Evidence of Through-Space Electrostatic Stabilization of the Incipient Oxocarbenium Ion by an Axially Oriented Electronegative Substituent During Glycopyranoside Acetolysis", The Journal of Organic Chemistry, 62 (22): 7597–7604, doi:10.1021/jo970677d
- ↑ Vollhardt; Shore (2009). Organic Chemistry. New York, NY: W. H. Freeman and Co.
- 1 2 3 Harmata, M.; Rashatasakhon, P. (2003), "Cycloaddition reactions of vinyl oxocarbenium ions", Tetrahedron, 59: 2371–2395, doi:10.1016/s0040-4020(03)00253-9
- ↑ Kanwar, S.; Trehan, S. (2005), "Acetate aldol reactions of chiral oxocarbenium ions", Tetrahedron Letters, 46 (8): 1329–1332, doi:10.1016/j.tetlet.2004.12.116
- ↑ Carrick, J. D.; Jennings, M. P. (2009), "An Efficient Formal Synthesis of (−)-Clavosolide a Featuring a "Mismatched" Stereoselective Oxocarbenium Reduction", Organic Letters, 11 (3): 769–772, doi:10.1021/ol8028302
- ↑ Martinez-Solorio, D.; Jennings, M. P. (2010), "Formal Synthesis of (−)-Neopeltolide Featuring a Highly Stereoselective Oxocarbenium Formation/Reduction Sequence", The Journal of Organic Chemistry, 75 (12): 4095–4104, doi:10.1021/jo100443h
- ↑ Maton, Anthea; Jean Hopkins; Charles William McLaughlin; Susan Johnson; Maryanna Quon Warner; David LaHart; Jill D. Wright (1993). Human Biology and Health. Englewood Cliffs, New Jersey, USA: Prentice Hall. pp. 52–59. ISBN 0-13-981176-1.
- ↑ Muller-Steffner, H. M.; Augustin, A.; Schuber, F. (1996), "Mechanism of Cyclization of Pyridine Nucleotides by Bovine Spleen NAD+ Glycohydrolase", Journal of Biological Chemistry, 271 (39): 23967–23972, doi:10.1074/jbc.271.39.23967
- ↑ Zechel, D.L.; Withers, S.G. (2000). "Glycosidase Mechanisms: Anatomy of a Finely Tuned Catalyst". Accounts of Chemical Research. 33 (1): 11–18. doi:10.1021/ar970172. ISSN 0001-4842.