Parastremmatic dwarfism
| Parastremmatic dwarfism | |
|---|---|
| Classification and external resources | |
| OMIM | 168400 |
| DiseasesDB | 33541 |
Parastremmatic dwarfism is a rare bone disease that features severe dwarfism, thoracic kyphosis (a type of scoliosis that affects the upper back), a distortion and twisting of the limbs, contractures of the large joints, malformations of the vertebrae and pelvis, and incontinence. The disease was first reported in 1970 by Leonard Langer and associates; they used the term parastremmatic from the Greek parastremma, or distorted limbs, to describe it. On X-rays, the disease is distinguished by a "flocky" or lace-like appearance to the bones.[1] The disease is congenital, which means it is apparent at birth. It is caused by a mutation in the TRPV4 gene, located on chromosome 12 in humans. The disease is inherited in an autosomal dominant manner.[1][2][3]
Characteristics
Parastremmatic dwarfism is apparent at birth, with affected infants usually being described as "stiff", or as "twisted dwarfs" when the skeletal deformities and appearance of dwarfism further present themselves. Skeletal deformities usually develop in the sixth to twelfth month of an infant's life. The deformities may be attributed to osteomalacia, a lack of bone mineralization.
Cause and genetics
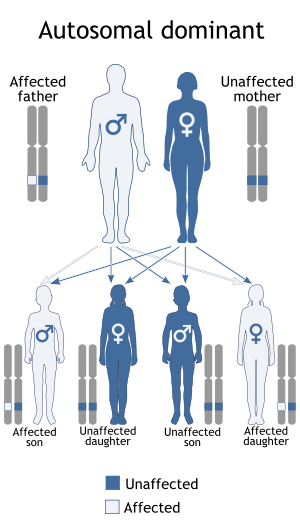
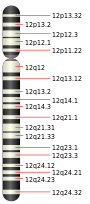
Parastremmatic dwarfism is caused by a missense mutation (where one amino acid is replaced by another in a gene sequence) in the TRPV4 gene,[3] located on the long arm of human chromosome 12, at 12q24.11.[4] The mutation is in exon 11 of the gene, and is labelled R594H; this means that the codon (the code for an amino acid molecule) for arginine was erroneously substituted by a codon for histidine at position 594 in that exon. This same mutation in the TRVP4 gene is known to cause the Kozlowski type of spondylometaphyseal dysplasia.[3][5]
Parastremmatic dwarfism is inherited in an autosomal dominant manner,[6] which means that the defective gene responsible for the disease is located on an autosome (chromosome 12 is an autosome), and one copy of the defective gene is sufficient to cause the disorder when inherited from a parent who also has the disorder.[1]
References
- 1 2 3 Horan, F.; Beighton, P. (Aug 1976). "Parastremmatic dwarfism" (Free full text). The Journal of Bone and Joint Surgery. British volume. 58 (3): 343–346. PMID 956253.
- ↑ Langer, L. O.; Petersen, D.; Spranger, J. (Nov 1970). "An unusual bone dysplasia: Parastremmatic dwarfism" (Free full text). The American Journal of Roentgenology, Radium Therapy, and Nuclear Medicine. 110 (3): 550–560. PMID 4992387.
- 1 2 3 Nishimura, G.; Dai, J.; Lausch, E.; Unger, S.; Megarbané, A.; Kitoh, H.; Kim, O. H.; Cho, T. J.; Bedeschi, F.; Benedicenti, F.; Mendoza-Londono, R.; Silengo, M.; Schmidt-Rimpler, M.; Spranger, J.; Zabel, B.; Ikegawa, S.; Superti-Furga, A. (Jun 2010). "Spondylo-epiphyseal dysplasia, Maroteaux type (pseudo-Morquio syndrome type 2), and parastremmatic dysplasia are caused by TRPV4 mutations". American Journal of Medical Genetics Part A. 152A (6): 1443–1449. doi:10.1002/ajmg.a.33414. PMID 20503319.
- ↑ Online Mendelian Inheritance in Man (OMIM) Parastremmatic dwarfism -168400 Retrieved 11-19-2011.
- ↑ Online Mendelian Inheritance in Man (OMIM) Transient Receptor Potential Cation Channel, Subfamily V, Member 4; TRPV4 -605427#0003 Retrieved 11-19-2011.
- ↑ Canepa, G.; Maroteaux, P.; Pietrogrande, V. (2000). Dysmorphic Syndromes and Constitutional Disease of the Skeleton. PICCIN. pp. 1421–1424. ISBN 8829915025.