Proteostasis
Proteostasis, a portmanteau of the words protein and homeostasis, is the concept that there are competing and integrated biological pathways within cells that control the biogenesis, folding, trafficking and degradation of proteins present within and outside the cell.[1][2] The concept of proteostasis maintenance is central to understanding the cause of diseases associated with excessive protein misfolding and degradation leading to loss-of-function phenotypes,[3] as well as aggregation-associated degenerative disorders.[4] Therefore, adapting proteostasis should enable the restoration of proteostasis once its loss leads to pathology. Cellular proteostasis is key to ensuring successful development, healthy aging, resistance to environmental stresses, and to minimize homeostasis perturbations by pathogens such as viruses.[2] Mechanisms by which proteostasis is ensured include regulated protein translation, chaperone assisted protein folding and protein degradation pathways. Adjusting each of these mechanisms to the demand for proteins is essential to maintain all cellular functions relying on a correctly folded proteome.
Mechanisms of proteostasis
The roles of the ribosome In proteostasis
One of the first points of regulation for proteostasis is during translation. This is accomplished via the structure of the ribosome, a complex central to translation. These two characteristics shape the way the protein folds and influences the proteins future interactions. The synthesis of a new peptide chain using the ribosome is very slow and the ribosome can even be stalled when it encounters a rare codon, a codon found at low concentrations in the cell.[5] These pauses provide an opportunity for an individual protein domain to have the necessary time to become folded before the production of following domains. This facilitates the correct folding of multi-domain proteins.[5] The newly synthesized peptide chain exits the ribosome into the cellular environment through the narrow ribosome exit channel (width: 10Å to 20Å, length 80Å).[5] Due to space restriction in the exit channel the nascent chain already forms secondary and limited tertiary structures. For example, an alpha helix is one such structural property that is commonly induced in this exit channel.[6] At the same time the exit channel also prevents premature folding by impeding large scale interactions within the peptide chain which would require more space.
Molecular chaperones and post-translational maintenance in proteostasis
In order to maintain protein homeostasis post-translationally, the cell makes use of molecular chaperones and/or chaperonins, which aid in the assembly or disassembly of proteins.[7] They recognize exposed segments of hydrophobic amino acids in the nascent peptide chain and then work to promote the proper formation of noncovalent interactions that lead to the desired folded state.[7] Chaperones begin to assist in protein folding as soon as a nascent chain longer than 60 amino acids emerges from the ribosome exit channel.[8] One of the most studied ribosome binding chaperones is trigger factor. Trigger Factor works to stabilize the peptide, promotes its folding, prevents aggregation, and promotes refolding of denatured model substrates.[9] Trigger factor not only directly works to properly fold the protein but also recruits other chaperones to the ribosome, such as Hsp70. Hsp70 surrounds an unfolded peptide chain, thereby preventing aggregation and promoting folding.,[7][8]
Chaperonins are a special class of chaperones that promote native state folding by cyclically encapsulating the peptide chain.[8] Chaperonins are divided into two groups. Group 1 chaperonins are commonly found in bacteria, chloroplasts, and mitochondria. Group 2 chaperonins are found in both the cytosol of eukaryotic cells as well as in archaea.[10] Group 2 chaperonins also contain an additional helical component which acts as a lid for the cylindrical protein chamber, unlike Group 1 which instead relies on an extra cochaperone to act as a lid. All chaperonins exhibit two states (open and closed), between which they can cycle. This cycling process is important during the folding of an individual polypeptide chain as it helps to avoid undesired interactions as well as to prevent the peptide from entering into kinetically trapped states.[10]
Regulating proteostasis by protein degradation
The third component of the proteostasis network is the protein degradation machinery. Protein degradation occurs in proteostasis when the cellular signals indicate the need to decrease overall cellular protein levels. The effects of protein degradation can be local, with the cell only experiencing effects from the loss of the degraded protein itself or widespread, with the entire protein landscape changing due to loss of other proteins’ interactions with the degraded protein.[6] Multiple substrates are targets for proteostatic degradation. These degradable substrates include nonfunctional protein fragments produced from ribosomal stalling during translation, misfolded or unfolded proteins, aggregated proteins, and proteins that are no longer needed to carry out cellular function. Several different pathways exist for carrying out these degradation processes. When proteins are determined to be unfolded or misfolded, they are typically degraded via the unfolded protein response (UPR) or ER associated degradation (ERAD). Substrates that are unfolded, misfolded, or no longer required for cellular function can also be ubiquitin tagged for degradation by ATP dependent proteases, such as the proteasome in eukaryotes or ClpXP in prokaryotes. Autophagy, or self engulfment, lysosomal targeting, and phagocytosis (engulfment of waste products by other cells) can also be used as proteostatic degradation mechanisms.[6]
Signaling events in proteostasis
Protein misfolding is detected by mechanisms that are specific for the cellular compartment in which they occur. Distinct surveillance mechanisms that respond unfolded protein have been characterized in the cytoplasm, ER and mitochondria. This response acts locally in a cell autonomous fashion but can also extend to intercellular signaling to protect the organism from anticipated proteotoxic stress.
Cell-autonomous stress responses
Cellular stress response pathways detect and alleviate proteotoxic stress which is triggered by imbalances in proteostasis. The cell-autonomous regulation occurs through direct detection of misfolded proteins or inhibition of pathway activation by sequestering activating components in response to heat shock. Cellular responses to this stress signaling include transcriptional activation of chaperone expression, increased efficiency in protein trafficking and protein degradation and translational reduction.
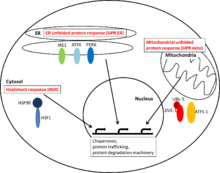
Cytosolic heat shock response
The cytosolic HSR is mainly mediated by the transcription factor family HSF (heat shock family). HSF is constitutively bound by Hsp90. Upon a proteotoxic stimulus Hsp90 is recruited away from HSF which can then bind to heat response elements in the DNA and upregulate gene expression of proteins involved in the maintenance of proteostasis.
ER unfolded protein response
The unfolded protein response in the endoplasmatic reticulum is activated by imbalances of unfolded proteins inside the ER and the proteins mediating protein homeostasis. Different “detectors” - such as IRE1, ATF6 and PERK - can recognize misfolded proteins in the ER and mediate transcriptional responses which help alleviate the effects of ER stress.
Mitochondrial unfolded protein response
The mitochondrial unfolded protein response detects imbalances in protein stoichiometry of mitochondrial proteins and misfolded proteins. The expression of mitochondrial chaperones is upregulated by the activation of the transcription factors ATF-1 and/or DVE-1 with UBL-5.
Systemic stress signaling
Stress responses can also be triggered in a non-cell autonomous fashion by intercellular communication. The stress that is sensed in one tissue could thereby be communicated to other tissues to protect the proteome of the organism or to regulate proteostasis systemically. Cell non-autonomous activation can occur for all three stress responses.
Work on the model organism C. elegans has shown that neurons play a role in this intercellular communication of cytosolic HSR. Stress induced in the neurons of the worm can in the long run protect other tissues such as muscle and intestinal cells from chronic proteotoxicity. Similarly ER and mitochondrial UPR in neurons are relayed to intestinal cells . These systemic responses have been implicated in mediating not only systemic proteostasis but also influence organismal aging.[11]
Diseases of proteostasis
Proteostasis and diseases of protein folding
Dysfunction in proteostasis can arise from errors in or misregulation of protein folding. The classic examples are missense mutations and deletions that change the thermodynamic and kinetic parameters for the protein folding process.[1] These mutations are often inherited and range in phenotypic severity from having no noticeable effect to embryonic lethality. Disease develops when these mutations render a protein significantly more susceptible to misfolding, aggregation, and degradation. If these effects only alter the mutated protein, the negative consequences will only be local loss of function. However, if these mutations occur in a chaperone or a protein that interacts with many other proteins, dramatic global alterations in the proteostasis boundary will occur. Examples of diseases resulting from proteostatic changes from errors in protein folding include cystic fibrosis, Huntington’s disease, Alzheimer’s disease, lysosomal storage disorders, and others.[12]
The role of model systems in the elucidation of protein-misfolding diseases
Small animal model systems have been and continue to be instrumental in the identification of functional mechanisms that safeguard proteostasis. Model systems of diverse misfolding-prone disease proteins have so far revealed numerous chaperone and co-chaperone modifiers of proteotoxicity.[13]
Proteostasis and cancer
The unregulated cell division that marks cancer development requires increased protein synthesis for cancer cell function and survival. This increased protein synthesis is typically seen in proteins that modulate cell metabolism and growth processes. Cancer cells sometimes susceptible to drugs that inhibit chaperones and disrupt proteostasis, such as Hsp90 inhibitors or proteasome inhibitors.[1]
Proteostasis and obesity
A hallmark of cellular proteostatic networks is their ability to adapt to stress via protein regulation. Metabolic disease, such as that associated with obesity, alters the ability of cellular proteostasis networks adapt to stress, often with detrimental health effects. For example, when insulin production exceeds the cell’s insulin secretion capacity, proteostatic collapse occurs and chaperone production is severely impaired. This disruption leads to the disease symptoms exhibited in individuals with diabetes.[1]
Proteostasis and aging
Over time, the proteostasis network becomes burdened with proteins modified by reactive oxygen species and metabolites that induce oxidative damage.[1] These byproducts can react with cellular proteins to cause misfolding and aggregation (especially in nondividing cells like neurons). This risk is particularly high for intrinsically disordered proteins. The IGFR-1 pathway has been shown in C. elegans to protect against these harmful aggregates, and some experimental work has suggested that upregulation of insulin growth factor receptor 1 (IGFR-1) may stabilize proteostatic network and prevent detrimental effects of aging.[1] Expression of the chaperome, the ensemble of chaperones and co-chaperones that interact in a complex network of molecular folding machines to regulate proteome function, is dramatically repressed in human aging brains and in the brains of patients with neurodegenerative diseases. Functional assays in C. elegant and human cells have identified a conserved chaperome sub-network of 16 chaperone genes, corresponding to 28 human orthologs as a proteostasis safeguard in aging and age-onset neurodegenerative disease. [14]
Pharmacologic intervention in proteostasis
There are two main approaches that have been used for therapeutic development targeting the proteostatic network: pharmacologic chaperones and proteostasis regulators. The principle behind designing pharmacologic chaperones for intervention in diseases of proteostasis is to design small molecules that stabilize borderline proteins. Previously, this approach has been used to target and stabilize G-protein coupled receptors, neurotransmitter receptors, glycosidases, lysosomal storage proteins, and the mutant CFTR protein that causes cystic fibrosis.[1]
The principle behind proteostasis regulators is similar, but seeks to stabilize the proteostatic boundary on a widespread scale. For example, some proteostasis regulators target chaperones such as heat shock proteins for stabilization. It has been suggested that this approach could even be applied prophylactically, such as upregulating certain protective pathways before experiencing an anticipated severe cellular stress. One theoretical mechanism for this approach includes upregulating the unfolded protein response to rescue proteins from degradation during cellular stress.[1]
References
- 1 2 3 4 5 6 7 8 Powers, E.T.; Morimoto, R.I.; Dillin, A.; Kelly, J.W.; Balch, W.E. (2009). "Biological and Chemical Approaches to Diseases of Proteostasis Deficiency". Annu. Rev. Biochem. 78: 959–91. doi:10.1146/annurev.biochem.052308.114844. PMID 19298183.
- 1 2 Balch WE, Morimoto RI, Dillin A, Kelly JW (Feb 2008). "Adapting proteostasis for disease intervention". Science. 319: 916–919. doi:10.1126/science.1141448. PMID 18276881.
- ↑ Mu, T-W.; Ong, D.S.T.; Wang, Y-J; Balch, W. E.; Yates, J.R.; Segatori, L.; Kelly, J.W. (2008). "Chemical and Biological Approaches Synergize to Ameliorate Protein-Folding Diseases". Cell. 134: 769–781. doi:10.1016/j.cell.2008.06.037. PMID 18775310.
- ↑ Cohen, E., Paulsson, J. F., Blinder, P., Burstyn-Cohen, T., Du, D., Estepa, G., Adame, A., Pham, H. M., Holzenberger, M., Kelly, J. W., Masliah, E. & Dillin, A. (2009). "Reduced IGF-1 signaling delays age-associated proteotoxicity in mice". Cell. 139: 1157–69. doi:10.1016/j.cell.2009.11.014. PMID 20005808.
- 1 2 3 Cavagnero, S. & Fedyukina, D. V. (March 2011). "Protein Folding at the Exit Tunnel". Annual Review of Biophysics. 40: 337–359. doi:10.1146/annurev-biophys-042910-155338. PMID 21370971.
- 1 2 3 Bustamante, C. J., et.al. (2014). "Mechanisms of Cellular Proteostasis: Insights from Single-Molecule Approaches". Annual Review of Biophysics. 43: 119–140. doi:10.1146/annurev-biophys-051013-022811. PMID 24895851.
- 1 2 3 Ye, K., et.al. (2013). "Molecular Chaperone Functions in Protein Folding and Proteostasis". Annual Review of Biophysics. 82: 323–355. doi:10.1146/annurev-biochem-060208-092442. PMID 23746257.
- 1 2 3 Vabulas, M. R. et. al. (2010). "Protein Folding in the Cytoplasm and the Heat Shock Response". Cold Spring Harb Perspect Biology. 2: 1–18. doi:10.1101/cshperspect.a004390. PMID 21123396.
- ↑ Hoffman, A. (June 2010). "Structure and function of the molecular chaperone Trigger Factor". Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 1803: 650–661. doi:10.1016/j.bbamcr.2010.01.017. PMID 20132842.
- 1 2 Yébenes, H., et. al. (Aug 2011). "Chaperonins: two rings for folding". Trends Biochem Sci. 36: 424–432. doi:10.1016/j.tibs.2011.05.003. PMID 21723731.
- ↑ Taylor RC, et al. (2014). "Systemic stress signalling: understanding the cell non-autonomous control of proteostasis". Nature Reviews Molecular Cell Biology. 15: 506–14. doi:10.1038/nrm3752. PMID 24556842.
- ↑ Hipp MS, et al. (2014). "Proteostasis impairment in protein-misfolding and aggregation diseases". Trends Cell Biol. 24: 211–217. doi:10.1016/j.tcb.2014.05.003. PMID 24946960.
- ↑ Brehme M, Voisine C (2016). "Model systems of protein-misfolding diseases reveal chaperone modifiers of proteotoxicity". Dis Model Mech. 9. doi:10.1242/dmm.024703. PMID 27491084.
- ↑ Brehme M, et al. (2014). "A conserved chaperome sub-network safeguards protein homeostasis in aging and neurodegenerative disease". Cell Rep. 9: 1135–1150. doi:10.1016/j.celrep.2014.09.042. PMID 25437566.
External links
Proteostasis: where do you work on the network? - Introduction to Proteostasis by Enzo Life Sciences
- Proteostasis Therapeutics Biotechnology company developing proteostasis regulators