White–Chen catalyst
 | |
| Identifiers | |
|---|---|
| 959395-10-9 | |
| ECHA InfoCard | 100.224.119 |
| Properties | |
| C24H32F12FeN6Sb2 | |
| Molar mass | 931.91 g·mol−1 |
| Appearance | Purple powder |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| Infobox references | |
The White–Chen catalyst is an Iron-based coordination complex named after Professor M. Christina White and her graduate student Mark S. Chen. The catalyst is used along with hydrogen peroxide and acetic acid additive to oxidize aliphatic sp3 C-H bonds in organic synthesis. The catalyst is the first to allow for preparative and predictable aliphatic C–H oxidations over a broad range of organic substrates.[1][2][3][4] Oxidations with the catalyst have proven to be remarkably predictable based on sterics, electronics, and stereoelectronics allowing for aliphatic C–H bonds to be thought of as a functional group in the streamlining of organic synthesis.
Electronic selectivity
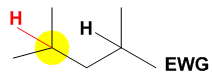
In the case where an electron withdrawing group (EWG) is present in the substrate the highly electrophilic catalyst will oxidize the more electron-rich C–H bond that is most remote from the EWG. In the case above the C–H bond shaded yellow is further from the electron withdrawing group and therefore has a higher electron density than the one not shaded yellow. The yellow shaded C–H bond is therefore the primary site for oxidation by the catalyst.
- Example of Electronic Selectivity
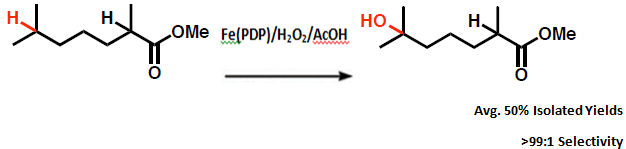
The reaction selectivity is highly influenced by electronics due to the highly electron withdrawing ester group present in the substrate. For that reason the reaction proceeds with oxidation at the tertiary C–H bond most remote from the ester with reaction yields of greater than 50% and selectivities of >99:1.[5] Electronically guided site-selectivity is also observed for secondary sites, affording yields of mono-oxidized products of 50% or greater.[6]
Steric selectivity
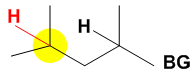
In the case where a bulky group, denoted at the right by BG, is present near multiple aliphatic C-H groups that are electronically equivalent, the bulky White–Chen catalyst will target the less sterically hindered C-H bond. In the case at the right there is a large bulky group in close proximity to one of two aliphatic C-H bonds. The C-H bond shaded in yellow is further from the bulky group, is less sterically hindered, and will therefore be the site of oxidation by the catalyst in this case. The bulky nature of the White–Chen catalyst allows product predictability based on steric properties of the starting materials given that electronic properties are equivalent.
- Example of steric selectivity
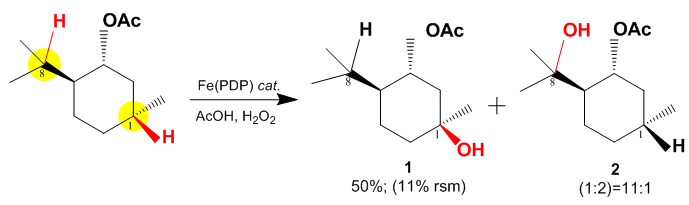
In the above case there are two electronically favored C–H sites in the substrate, both highlighted in yellow (Carbons 1 and 8), for oxidation with the White–Chen catalyst. Due to the large steric hindrance at Carbon 8 the reaction proceeds by primarily oxidizing Carbon 1. The reaction proceeds with 50% yield and with a selectivity of 11:1 for C-1 vs. C-8.[5]
Stereoelectronic Selectivity
In the case where there is an electron activating group (EAG) near aliphatic C–H bonds, the EAG will donate electron density to the adjacent C–H bonds to promote oxidation at that site. In the case at the right the cyclopropane EAG activates the aliphatic C–H bonds adjacent to it via hyperconjugation (oxygen and nitrogen heteroatoms can also do this). The bonds highlighted in red will undergo oxidation preferentially to other C—H bonds in the molecule. The observed product is shown on the right and consists of a carbonyl in place of the two aliphatic C–H bonds. Other oxidants such as TFDO and DMDO have also been shown to oxidize aliphatic C—H bonds selectively via stereoelectronic activation.[2][7]
- Example of stereoelectronic selectivity

The above reaction is an example of the stereoelectronic selectivity-based strain relief (alleviation of 1,3-diaxial interactions, torsional strain, etc.). In this case torsional strain between the bulky tert-butyl group and the adjacent methylenes is relieved upon oxidation at C3. The carbon highlighted in yellow is the main site of oxidation yielding the product at the right with a reaction yield of 59%. The minor oxidation product at C4 was formed in 12%.[6]
Directing group selectivity

In the case where a directing group, denoted at the right by DG, is present near multiple aliphatic C-H groups the catalyst will oxidize the C-H bond closest to the directing group.[8][9][10] Thus far, only carboxylic acid functional groups have been shown to be effective and when present, the AcOH additive does not need to be added to the reaction. In the case at the right the C–H bond highlighted in yellow is in close proximity to the directing group and would be the expected site for oxidation in this substrate.
- Example of directing group selectivity
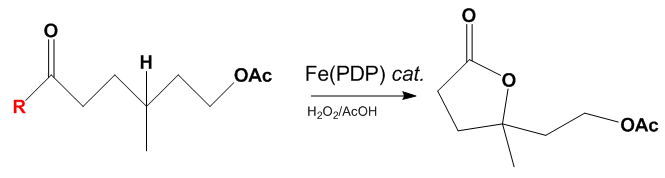
The above figure shows the effects of a carboxylic acid directing group on the overall reactivity of the reaction.[11] The substrate on the left is the starting material and when the R group is an ester the oxidation takes place with low yields of about 26% due to electronic deactivation of the proximal C—H bond. When the R group is a carboxylic acid the yield increases to 50%. In general the White Chen catalyst will bind to a carboxylic acid groups and this can be used to override negative electronic, steric, and stereoelectronic effects within a molecule.
- Example of directing group selectivity

The above figure shows a carboxylic acid directed diastereoselective lactonization of tetrahydrogibberellic acid via a C-H oxidation to form a butyrolactone derivative.[5]
Combined effects in complex molecules
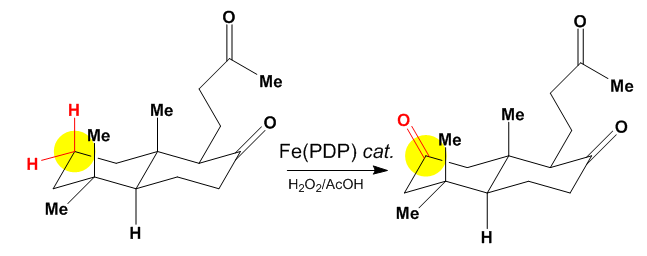
The above figure shows oxidation at the least sterically hindered and electronically/stereo-electronically activated site. The White–Chen catalyst relies on the constructive combination of inherent electronic, steric, and stereoelectronic factors within a substrate to favor one site of oxidation. When these factors combine productively, the reaction proceeds with great predictability and an isolated yields of 50% or greater.[6]
Mechanism
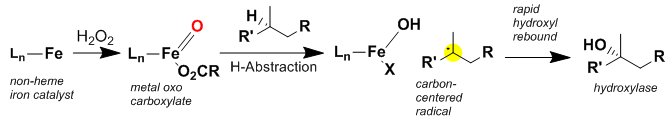
The above figure illustrates the proposed mechanism for oxidation by the White–Chen catalyst. The reaction proceeds in conjunction with hydrogen peroxide oxidant and acetic acid. The catalyst quickly reacts with hydrogen peroxide, believed to be catalyzed by the acetic acid, and forms and iron-oxo intermediate.[12] The iron-oxo intermediate is then thought to abstract a hydrogen on the carbon to be oxidized to generate a short-lived carbon centered radical. The iron-hydroxide then quickly rebounds onto the carbon centered radical to form the oxidized compound. Recently, direct evidence was obtained for a carbon-centered radical intermediate in this reaction using a Taxane-based radical trap.[13] It is important to note that these reactions (both directed and non-directed) proceed with complete retention of stereochemistry.
References
- ↑ Barton, D.H.R; Csuha, E; Ozbalik, N (1990). Tetrahedron. 46: 3743–3752. doi:10.1016/s0040-4020(01)90510-1. Missing or empty
|title=(help) - 1 2 Mello, R; Fiorentino, M.; Fusco, C; Curci,R (1989). J. Am. Chem. Soc. 111: 6749. doi:10.1021/ja00199a039. Missing or empty
|title=(help) - ↑ Kim, C; Chen, K.J.; Kim, J.; Que Jr., L. (1997). J. Am. Chem. Soc. 119: 5964. doi:10.1021/ja9642572. Missing or empty
|title=(help) - ↑ Groves, J.T.; Nemo, T.E.; Myers, R.S. (1979). J. Am. Chem. Soc. 101: 1032. doi:10.1021/ja00498a040. Missing or empty
|title=(help) - 1 2 3 Chen, Mark S.; White, M. Christina (2007). "A Predictably Selective Aliphatic C–H Oxidation Reaction for Complex Molecule Synthesis". Science. 318. doi:10.1126/science.1148597.
- 1 2 3 Chen, Mark S.; White, M. Christina (2010). "Combined Effects on Selectivity in Fe-Catalyzed Methylene Oxidation". Science. 327: 566. doi:10.1126/science.1183602.
- ↑ Wender, P.A.; Hilinski, M.K.; Mayneg, A.V.W (2005). Organic Letters. 7: 79. Missing or empty
|title=(help) - ↑ Desai, L.V.; Hull, K.L.; Sandford, M.S. (2004). J. Am. Chem. Soc. 126: 9542. doi:10.1021/ja046831c. Missing or empty
|title=(help) - ↑ Giri, R.; Chen, X.; Yu, J-Q (2005). Angew. Chem. Int. Ed. 44: 2112. doi:10.1002/anie.200462884. Missing or empty
|title=(help) - ↑ Simmons, E.M.; Hartwig, J.F. (2012). Nature. 483: 70. doi:10.1038/nature10785. Missing or empty
|title=(help) - ↑ Bigi, M.A.; Reed, S.A.; White, M.C (2012). J. Am. Chem. Soc. 134: 9721–9726. doi:10.1021/ja301685r. Missing or empty
|title=(help) - ↑ Chen,K (2001). "Stereospecific alkane hydroxylation by non-heme iron catalysts: mechanistic evidence for an Fe=O active species". J. Am. Chem. Soc. 123: 6327–6337. doi:10.1021/ja010310x.
- ↑ Bigi, M.A.; Reed, S.A.; White, M.C. (2011). "Diverting non-heme iron catalyzed aliphatic C-H hydroxylations towards desaturations". Nature Chemistry. 3: 218. doi:10.1038/NCHEM.967.
External links
- "Nature Article: Unlocking ketones". SciBX. 3 (6). doi:10.1038/scibx.2010.171.