Acquired non-inflammatory myopathy

Acquired non-inflammatory myopathy (ANIM) is a neurological disorder primarily affecting skeletal muscle, most commonly in the limbs of humans, resulting in a weakness or dysfunction in the muscle.[1] A myopathy refers to a problem or abnormality with the myofibrils, which compose muscle tissue. In general, non-inflammatory myopathies are a grouping of muscular diseases not induced by an autoimmune-mediated inflammatory pathway. These muscular diseases usually arise from a pathology within the muscle tissue itself rather than the nerves innervating that tissue. ANIM has a wide spectrum of causes which include drugs and toxins, nutritional imbalances, acquired metabolic dysfunctions such as an acquired defect in protein structure, and infections.[1]
Acquired non-inflammatory myopathy is a different diagnosis than inflammatory myopathy. Inflammatory myopathies are a direct result of some type of autoimmune mediated pathway whereas ANIM is not the result of a dysfunction of the immune system.[2] In addition, the cause of inflammatory myopathy is relatively unknown, whereas many causal agents for ANIM have been discovered which typically affect the structural integrity and function of the muscle fibers.[2]
Most myopathies are typically first diagnosed and classified as an idiopathic inflammatory myopathy.[2] However, a diagnosis of ANIM occurs when the cause of the myopathy is found to not arise from an autoimmune mechanism.[1]
Symptoms
Patients with acquired non-inflammatory myopathy typically experience weakness, cramping, stiffness, and tetany, most commonly in skeletal muscle surrounding the limbs and upper shoulder girdle.[1]
The most commonly reported symptoms are:
- Muscle fatigue[1]
- Pain[1]
- Muscle spasms and cramps
- Tingling
- Numbness
- Tetany[1]
- Loss of coordination and balance[1]
- Lack of fine and gross motor control[1]
- Muscular wasting and atrophy[1]
Epidemiology
Acquired noninflammatory myopathy can be caused by a variety of factors including metabolic abnormalities, drugs, nutritional deficiency, trauma, and upstream abnormalities resulting in decreased function. Two of the most common causes of ANIM are hyperthyroidism and excessive steroid use, while many drugs used to treat rheumatism are known to be inducing agents. Most cases of ANIM can be linked to drugs or dietary abnormalities.
Drug induced myopathy
It is not uncommon for drugs to damage muscle fibers. Particular families of drugs are known to induce myopathies on the molecular level, thus altering organelle function such as the mitochondria. Use of multiple drugs from these families in conjunction with one another can increase the risk of developing a myopathy.[3] Many of the drugs associated with inducing myopathies in patients are found in rheumatology practice.
Statins
- Prescribed statins for dyslipidemia are associated with muscle toxicity. Symptoms of this muscle toxicity include combinations of cramping, weakness, aching or tenderness; and are often experienced in the quadriceps, pectoral, biceps, low back, or abdominal region. Symptoms tend to worsen with muscle exercise, and often continue after a patient is removed from statin therapy.[1] Common types of myopathy due to statins include myalgia, myositis, and rhabdomyolysis. Statins induce myopathy by inhibiting protein synthesis within the muscle.[3] Statin therapy tends to not show any histopathological differences, and thus a biopsy does not reveal too much about the damage. Often, the damage is found within the mitochondria.[1]
Corticosteriods
- Corticosteriods often cause muscle weakness to some degree in patients. Symptoms are usually weakness of the proximal muscles, neck flexor, and in extreme cases, respiratory muscle weakness can also occur.[1] Corticosteriods have not only been found to cause some degree of muscle atrophy, but also a local or diffuse cell death. These side effects are more common in women than in men, for reasons that are unknown.[3] EMGs illicit a low amplitude motor potentials, and lack spontaneous electrical activity.[1]
Colchicine
- Patients who start taking colchicine, and who have compromised renal function, develop a myopathy that shows symptoms in proximal muscle weakness, distal sensory loss and areflexia. A muscle biopsy shows a vacuolar myopathy without significant cell death or inflammation.[1]
Chloroquine/Hydroxychloroquine
- Chloroquine/hydroxychloroquine prescriptions can cause a development of a progressively slow muscle weakness that begins in the low extremities, and moves to the upper limbs. Muscle enzymes are increased, commonly lactate dehydrogenase (LDH).[1]
(HMG-CoA) Reductase Inhibitors
- (HMG-CoA) reductase inhibitors are commonly used in the clinic to reduce lipid levels in patients. This drug is known to have toxic effects on myofibrils, resulting in muscle pain and tenderness. Biopsies demonstrate more molecular damage and dysfunction within the mitochondria, higher lipid storage, and vibrant red myofibrils. These symptoms are commonly found in the proximal muscles of the body.[3]
Diet and Trauma Induced Myopathy
Many dietary factors and aberrations can induce ANIM. Chemical imbalances brought on by abnormal diets may either affect the muscle directly or induce abnormal functionality in upstream pathways.
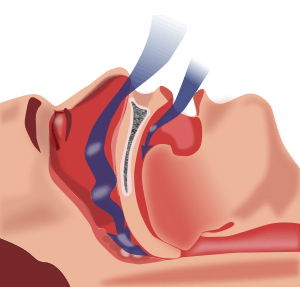
- Excess Iodine consumption, especially in the form of kelp, can induce Hyperthyroidism. Hyperthyroidism is one of the most common ways to acquire ANIM.[4] A hyperactive thyroid gland produces excessive amounts of hormones T3 and T4 leading to increased metabolism and increased sympathetic nervous system effects. The muscles exhibit a pathology similar to an overdose of epinephrine (commonly known as adrenaline). Patients with hyperthyroidism show weakness of shoulder girdle muscles in particular with this condition often being asymptomatic. More serious weakness of core and limb muscles may present.[4]
- A dietary deficiency of vitamin D is most commonly associated with osteoporosis, but can cause ANIM as well. Vitamin D induced ANIM is most commonly associated with sleep deprivation as it induces tonsillar and adenotonsillar hypertrophy, as well as weakens the airway muscles.[5] These changes induce sleep apnea and sleep disruption. Vitamin D induced ANM can also be associated with daytime impairment through this pathway.[5]
Trauma to any muscle is also a common cause for acute ANIM. This is due to muscular contusions and partial or complete loss of function for affected muscle groups.[2]
Diagnosis

A patient's history is one of the key factors in diagnosing acquired noninflammatory myopathy. The history is used not only to analyze the time frame with which the patient began to express symptoms, but to also see if the disease is within the patient's family's history, to check medication or drug use history, and to see if the patient has suffered any trauma due to illness or infection. Basic exams will test for where the muscle weakness is and how weak it is. This is performed by testing for proximal and distal muscle strength, as well as testing for any signs of neurogenic symptoms such as impaired sensation, deep tendon reflexes, and atrophy.[1]
If needed, more advanced equipment can be used to help determine whether a patient is suffering from ANIM. This includes:
- Measurement of serum levels of muscle enzymes[1]
- Electromyography (EMG)[1]
- Magnetic Resonance Imaging (MRI)[1]
- Muscle biopsy[1]
When examining the serum levels of muscle enzymes, the relative levels of creatine kinase, aldolase, aspartate aminotransferase, alanine aminotransferase, and lactate dehydrogenase are closely examined. Abnormal levels of these proteins are indicative of both inflammatory myopathy and ANIM.[1]
EMGs are particularly useful in locating the affected muscle groups, as well as determining the distribution of the myopathy throughout the cell. EMGs measure several indicators of myopathies such as:
- The spontaneous electrical movement from a single muscle fiber at rest,
- Measurement of a polyphasic, shorter amplitude, motor unit action potential during muscle stimulation,
- Determining that the muscle group cannot differentiate large motor plate stimulation from small motor plate stimulation involved in recruitment of muscle fibers.[1]
Magnetic Resonance Imaging will illicit edema in inflammatory patients, but it will most likely show nothing in patients with ANIM and if it does, it will show some atrophy.[1]
If an individual's ANIM is a result of a metabolite defect, then additional tests are required. These tests are directed at enzyme function at rest and during exercise, and enzyme intermediates. Molecular genetic testing is often used to determine if there was any predisposition to the expressed symptoms.[1]
Screening
During vigorous ischemic exercise, skeletal muscle functions aerobically, generating lactate and ammonia a coproduct of muscle myoadenylate deaminase (AMPD) activity. The forearm ischemic exercise test takes advantage of this physiology and has been standardized to screen for disorders of glycogen metabolism and AMPD deficiency. Patients with a glycogen storage disease manifest a normal increase in ammonia but no change from baseline of lactate, whereas in those with AMPD deficiency, lactate levels increase but ammonia levels do not. If ischemic exercise testing gives an abnormal result, enzyme analysis must be performed on muscle to confirm the putative deficiency state because false-positive results can occur.
Treatment

Treatment for acquired noninflammatory myopathy is directed towards resolution of the underlying condition, pain management, and muscle rehabilitation. Drug induced ANIMs can be reversed or improved by tapering off of the drugs and finding alternative care.[3] Hyperthyroidism induced ANIM can be treated through anti-thyroid drugs, surgery and not eating foods high in Iodine such as kelp. Treatment of the hyperthyroidism results in complete recovery of the myopathy.[4] ANIM caused by vitamin D deficiency can easily be resolved by taking vitamin supplements and increasing one's exposure to direct sunlight.
Pain can be managed through massaging affected areas and the use of nonsteroidal anti-inflammatory drugs (NSAIDs). Exercise, physical therapy, and occupational therapy can be used to rehabilitate affected muscle areas and resist the atrophy process.[2]
As with all myopathies, the use of walkers, canes, and braces can assist with the mobility of the afflicted individual.
Research direction
A diagnostic test for statin-associated auto-immune necrotizing myopathy will be available soon in order to differentiate between different types of myopathies during diagnosis. The presence of abnormal spontaneous electrical activity in the resting muscles indicates an irritable myopathy and is postulated to reflect the presence of an active necrotising myopathic process or unstable muscle membrane potential. However, this finding has poor sensitivity and specificity for predicting the presence of an inflammatory myopathy on biopsy. Further research into this spontaneous electrical activity will allow for a more accurate differential diagnosis between the different myopathies.[1]
Currently a muscle biopsy remains a critical test, unless the diagnosis can be secured by genetic testing. Genetic testing is a less invasive test and if it can be improved upon that would be ideal. Molecular genetic testing is now available for many of the more common metabolic myopathies and muscular dystrophies. These tests are costly and are thus best used to confirm rather than screen for a diagnosis of a specific myopathy. Due to the cost of these tests, they are best used to confirm rather than screen for a diagnosis of a specific myopathy. It is the hope of researchers that as these testing methods improve in function, both costs and access will become more manageable[3]
The increased study of muscle pathophysiology is of importance to researchers as it helps to better differentiate inflammatory versus non-inflammatory and to aim treatment as part of the differential diagnosis. Certainly classification schemes that better define the wide range of myopathies will help clinicians to gain a better understanding of how to think about these patients. Continued research efforts to help appreciate the pathophysiology will improve clinicians ability to administer the most appropriate therapy based on the particular variety of myopathy.[2]
The mechanism for myopathy in individuals with low vitamin D is not completely understood. A decreased availability of 250HD leads to mishandling of cellular calcium transport to the sarcoplasmic reticulum and mitochondria, and is associated with reduced actomyosin content of myofibrils.[5]
See also
References
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 Baer, Alan; Wortmann (May 2013). "Noninflammatory Myopathies". Rheuatic Disease Clinics of North America. 39 (2): 457–479. doi:10.1016/j.rdc.2013.02.006.
- 1 2 3 4 5 6 Castro, Christine; Gourley (April 2012). "Diagnosis and treatment of inflammatory myopathy: issues and management". Therapeutic Advances in Musculoskeletal Disease. 4 (2): 111–120. doi:10.1177/1759720x11425092.
- 1 2 3 4 5 6 Owczarek, Jacek; Magdalena (2005). "Drug-induced myopathies. An overview of the possible mechanisms". Pharmacol Rep. 57 (1): 23–34.
- 1 2 3 Wilkinson, Iain; Lennox, Graham (2005). Essential Neurology (4th ed.). Wiley-Blackwell. p. 171. ISBN 978-1-4051-1867-5.
- 1 2 3 McCarthy, D; Chesson (2013-09-25). "The link between vitamin D metabolism and sleep medicine". Sleep Med Rev.